Professional Documents
Culture Documents
Asuhan Keperawatan Thalesemia Pada Anak
Uploaded by
Q Chayanx KhamoeOriginal Title
Copyright
Available Formats
Share this document
Did you find this document useful?
Is this content inappropriate?
Report this DocumentCopyright:
Available Formats
Asuhan Keperawatan Thalesemia Pada Anak
Uploaded by
Q Chayanx KhamoeCopyright:
Available Formats
Keperawatan Anak II 1
BAB I
PENDAHULUAN
.1 Latar Belakang
Thalassemia adalah kelainan bawaan dari sintesis hemoglobin. Presentasi
klinisnya bervariasi dari asimtomatik sampai berat hingga mengancam jiwa. Dahulu
dinamakan sebagai Mediterannian anemia, diusulkan oleh Whipple, namun kurang
tepat karena sebenarnya kondisi ini dapat ditemukan di mana saja di seluruh dunia.
Seperti yang akan dijelaskan selanjutnya, beberapa tipe berbeda dari thalassemia
lebih endemik pada area geografis tertentu.
Pada tahun 1925, Thomas Cooley, seorang spesialis anak dari Detroit,
mendeskripsikan suatu tipe anemia berat pada anak-anak yang berasal dari Italia.
Beliau menemukan adanya nukleasi sel darah merah yang masif pada sapuan apus
darah tepi, yang mana awalnya beliau pikir sebagai anemia eritroblastik, suatu
keadaan yang disebutkan oleh von Jaksh sebelumnya. Namun tak lama kemudian,
Cooley menyadari bahwa eritroblastemia tidak spesifik dan esensial pada temuan ini
sehingga istilah anemia eritroblastik tidak dapat dipakai. Meskipun Cooley curiga
akan adanya pengaruh genetik dari kelainan ini, namun beliau gagal dalam
menginvestigasi orangtua sehat pada anak-anak yang mengidap kelainan ini.
Di Eropa, Riette mendeskripsikan mengenai adanya anemia mikrositik
hipokromik ringan yang tak terjelaskan pada anak-anak keturunan Italia pada tahun
yang sama saat Cooley melaporan adanya bentuk anemia berat yang akhirnya
dinamakan mengikutinya namanya. Sebagi tambahan, Wintrobe di Amerika Serikat
Asuhan keperawatan Thalesemia pada anak
Keperawatan Anak II 2
melaporkan adanya anemia ringan pada kedua orangtua dari anak yang mengidap
anemia Cooley. Anemia ini sangat mirip dengan kelainan yang ditemukan Riette.
Baru setelah itu anemia Cooley dinyatakan sebagai bentuk homozigot dari anemia
hipokromik mikrositik ringan yang dideskripsikan oleh Riette dan Wintrobe. Bentuk
anemia berat ini kemudian dilabelisasi sebagai thalassemia mayor dan bentuk
ringannya dinamakan sebagai thalassemia minor. Kata thalassemia berasal dari
bahasa Yunani yaitu thalassa yang berarti ‘laut’ (mengarah ke Mediterania), dan emia, yang
berarti berhubungan dengan darah.
.2 Tujuan
.2.1 Tujuan Umum
Untuk mengetahui tentang asuhan keperawatan pada klien dengan gastritis.
.2.2 Tujuan Khusus
1. Untuk mengetahui tentang konsep dasar teori tentang Thalesemia
pada anak.
2. Memberikan asuhan keperawatan pada klien dengan penyakit
Thalesemia asuhan keperawatan teoritis pada klien dengan penyakit
Thalesemia yang meliputi pengkajian, diagnosa dan intervensi
keperawatan.
.3 Manfaat
.3.1 Menambah pengetahuan dan keterampilan kelompok dalam menerapkan
asuhan keperawatan pada pasien penyakit gastritis.
.3.2 Menambah pengetahuan dan wawasan baik penulis maupun pembaca.
Asuhan keperawatan Thalesemia pada anak
Keperawatan Anak II 3
BAB II
LANDASAN TEORITIS
2.1 Konsep Teori Thalesemia
2.1.1 Definisi
Thalassemia adalah suatu penyakit congenital herediter yang diturunkan
secara autosom berdasarkan kelainan hemoglobin, di mana satu atau lebih
rantai polipeptida hemoglobin kurang atau tidak terbentuk sehingga
mengakibatkan terjadinya anemia hemolitik (Broyles, 1997). Dengan kata
lain, thalassemia merupakan penyakit anemia hemolitik, dimana terjadi
kerusakan sel darah di dalam pembuluh darah sehingga umur eritosit menjadi
pendek (kurang dari 120 hari). Penyebab kerusakan tersebut adalah Hb yang
tidak normal sebagai akibat dari gangguan dalam pembentukan jumlah rantai
globin atau struktur Hb.
Thalasemia merupakan penyakit anemia hemolitik herediter yang
diturunkan secara resesif, secara molekuler dibedakan menjadi thalasemia alfa
dan beta, sedangkan secara klinis dibedakan menjadi thalasemia mayor dan
minor ( Mansjoer, Kapita Selekta Kedokteran, 2000 : 497 )
Sindrom thalasemia adalah sekelompok penyakit atau keadaan herediter
dimana produksi satu atau lebih dari satu jenis rantai polipeptida terganggu
(Kosasih, 2001).
Asuhan keperawatan Thalesemia pada anak
Keperawatan Anak II 4
Thalasemia merupakan penyakit anemia hemofilia dimana terjadi
kerusakan sel darah merah di dalam pembuluh darah sehingga umur eritrosit
pendek (kurang dari 100 hari). (Ngastiyah, 1997).
Jadi Thalasemia adalah penyakit anemia hemolitik dimana terjadi kerusakan
sel darah merah (eritrosit) sehingga umur eritrosit pendek (kurang dari 100
hari), yang disebabkan oleh defesiensi produksi satu , yang diturunkan dari
keduaβ dan α atau lebih dari satu jenis rantai orang tua kepada anak-
anaknya secara resesif.
2.1.2 Etiologi
3 Faktor genetik yaitu perkawinan antara 2 heterozigot (carier) yang
menghasilkan keturunan Thalasemia (homozigot).
Talasemia disebabkan oleh delesi (hilangnya) satu gen penuh atau
sebagian dari gen (ini terdapat terutama pada talasemia -a) atau mutasi noktah
pada gen (terutama pada talasemia - b), kelainan itu menyebabkan
menurunnya sintesis rantai polipeptid yang menyusun globin.
Penyebab anemia pada talasemia bersifat primer dan sekunder. Primer
adalah berkurangnya sintesis HbA dan eritropoesis yang tidak efektif disertai
penghancuran sel-sel eritrosit intramedular. Sedangkan yang sekunder ialah
karena defisiensi asam folat, bertambahnya volume plasma intravaskular yang
mengakibatkan hemodilusi dan destruksi eritrosit oleh sistem
retikuloendotelial dalam limpa dan hati. Penelitian biomolekular
Asuhan keperawatan Thalesemia pada anak
Keperawatan Anak II 5
menunjukkan adanya mutasi DNA pada gen sehingga produksi rantai alfa
atau beta dari hemoglobin berkurang (Mansjoer, 2000).
3.1.1 Klasifikasi Thalessemia
Saat ini dikenal sejumlah besar sindrom thalasemia; masing-masing
melibatkan penurunan produksi satu atau lebih rantai globin, yang
membentuk bermacam-macam jenis Hb yang ditemukan pada sel darah
merah. Jenis yang paling penting dalam praktek klinis adalah sindrom yang
mempengaruhi baik atau sintesis rantai α maupun β.
Thalassemia-α
Anemia mikrositik yang disebabkan oleh defisiensi sintesis globin-α
banyak ditemukan di Afrika, negara di daerah Mediterania, dan sebagian
besar Asia. Delesi gen globin-α menyebabkan sebagian besar kelainan ini.
Terdapat empat gen globin-α pada individu normal, dan empat bentuk
thalassemia-α yang berbeda telah diketahui sesuai dengan delesi satu, dua,
tiga, dan semua empat gen ini
Tabel 1. Thalassemia-α
Genotip Jumlah gen α Presentasi Klinis Hemoglobin Elektroforesis
Saat Lahir > 6 bulan
αα/αα 4 Normal N N
-α/αα 3 Silent carrier 0-3 % Hb Barts N
--/αα atau –α/-α 2 Trait thal-α 2-10% Hb Barts N
--/-α 1 Penyakit Hb H 15-30% Hb Bart Hb H
--/-- 0 Hydrops fetalis >75% Hb Bart -
Ket : N = hasil normal, Hb = hemoglobin, Hb Bart’s = γ4, HbH = β4
1. Silent carrier thalassemia-α
Asuhan keperawatan Thalesemia pada anak
Keperawatan Anak II 6
o Merupakan tipe thalassemia subklinik yang paling umum, biasanya
ditemukan secara kebetulan diantara populasi, seringnya pada etnik Afro-
Amerika. Seperti telah dijelaskan sebelumnya, terdapat 2 gen α yang terletak
pada kromosom 16.
o Pada tipe silent carrier, salah satu gen α pada kromosom 16 menghilang,
menyisakan hanya 3 dari 4 gen tersebut. Penderita sehat secara hematologis,
hanya ditemukan adanya jumlah eritrosit (sel darah merah) yang rendah
dalam beberapa pemeriksaan.
o Pada tipe ini, diagnosis tidak dapat dipastikan dengan pemeriksaan
elektroforesis Hb, sehingga harus dilakukan tes lain yang lebih canggih. Bisa
juga dicari akan adanya kelainan hematologi pada anggota keluarga
( misalnya orangtua) untuk mendukung diagnosis. Pemeriksaan darah lengkap
pada salah satu orangtua yang menunjukkan adanya hipokromia dan
mikrositosis tanpa penyebab yang jelas merupakan bukti yang cukup kuat
menuju diagnosis thalasemia.
2. Trait thalassemia-α
o Trait ini dikarakterisasi dengan anemia ringan dan jumlah sel darah
merah yang rendah. Kondisi ini disebabkan oleh hilangnya 2 gen α pada satu
kromosom 16 atau satu gen α pada masing-masing kromosom. Kelainan ini
sering ditemukan di Asia Tenggara, subbenua India, dan Timur Tengah.
Asuhan keperawatan Thalesemia pada anak
Keperawatan Anak II 7
o Pada bayi baru lahir yang terkena, sejumlah kecil Hb Barts (γ4) dapat
ditemukan pada elektroforesis Hb. Lewat umur satu bulan, Hb Barts tidak
terlihat lagi, dan kadar Hb A2 dan HbF secara khas normal.
3. Penyakit Hb H
o Kelainan disebabkan oleh hilangnya 3 gen globin α, merepresentasikan
thalassemia-α intermedia, dengan anemia sedang sampai berat, splenomegali,
ikterus, dan jumlah sel darah merah yang abnormal. Pada sediaan apus darah
tepi yang diwarnai dengan pewarnaan supravital akan tampak sel-sel darah
merah yang diinklusi oleh rantai tetramer β (Hb H) yang tidak stabil dan
terpresipitasi di dalam eritrosit, sehingga menampilkan gambaran golf ball.
Badan inklusi ini dinamakan sebagai Heinz bodies.
4. Thalassemia-α mayor
o Bentuk thalassemia yang paling berat, disebabkan oleh delesi semua gen
globin-α, disertai dengan tidak ada sintesis rantai α sama sekali.
o Karena Hb F, Hb A, dan Hb A2 semuanya mengandung rantai α, maka
tidak satupun dari Hb ini terbentuk. Hb Barts (γ4) mendominasi pada bayi
yang menderita, dan karena γ4 memiliki afinitas oksigen yang tinggi, maka
bayi-bayi itu mengalami hipoksia berat. Eritrositnya juga mengandung
sejumlah kecil Hb embrional normal (Hb Portland = ζ2γ2), yang berfungsi
sebagai pengangkut oksigen.
o Kebanyakan dari bayi-bayi ini lahir mati, dan kebanyakan dari bayi yang
lahir hidup meninggal dalam waktu beberapa jam. Bayi ini sangat hidropik,
Asuhan keperawatan Thalesemia pada anak
Keperawatan Anak II 8
dengan gagal jantung kongestif dan edema anasarka berat. Yang dapat hidup
dengan manajemen neonatus agresif juga nantinya akan sangat bergantung
dengan transfusi.
Thalassemia-β
Sama dengan thalassemia-α, dikenal beberapa bentuk klinis dari
thalassemia-β; antara lain :
1. Silent carrier thalassemia-β
a. Penderita tipe ini biasanya asimtomatik, hanya ditemukan nilai eritrosit
yang rendah. Mutasi yang terjadi sangat ringan, dan merepresentasikan suatu
thalassemia-β+.
b. Bentuk silent carrier thalassemia-β tidak menimbulkan kelainan yang
dapat diidentifikasi pada individu heterozigot, tetapi gen untuk keadaan ini,
jika diwariskan bersama-sama dengan gen untuk thalassemia-β°,
menghasilkan sindrom thalassemia intermedia.
2. Trait thalassemia-β
a. Penderita mengalami anemia ringan, nilai eritrosit abnormal, dan
elektroforesis Hb abnormal dimana didapatkan peningkatan jumlah Hb A2,
Hb F, atau keduanya
b. Individu dengan ciri (trait) thalassemia sering didiagnosis salah sebagai
anemia defisiensi besi dan mungkin diberi terapi yang tidak tepat dengan
Asuhan keperawatan Thalesemia pada anak
Keperawatan Anak II 9
preparat besi selama waktu yang panjang. Lebih dari 90% individu dengan
trait thalassemia-β mempunyai peningkatan Hb-A2 yang berarti (3,4%-7%).
Kira-kira 50% individu ini juga mempunyai sedikit kenaikan HbF, sekitar 2-
6%. Pada sekelompok kecil kasus, yang benar-benar khas, dijumpai Hb A2
normal dengan kadar HbF berkisar dari 5% sampai 15%, yang mewakili
thalassemia tipe δβ.
3. Thalassemia-β yang terkait dengan variasi struktural rantai β
a. Presentasi klinisnya bervariasi dari seringan thalassemia media hingga
seberat thalassemia-β mayor
b. Ekspresi gen homozigot thalassemia (β+) menghasilkan sindrom mirip
anemia Cooley yang tidak terlalu berat (thalassemia intermedia). Deformitas
skelet dan hepatosplenomegali timbul pada penderita ini, tetapi kadar Hb
mereka biasanya bertahan pada 6-8 gr/dL tanpa transfusi.
c. Kebanyakan bentuk thalassemia-β heterozigot terkait dengan anemia
ringan. Kadar Hb khas sekitar 2-3 gr/dL lebih rendah dari nilai normal
menurut umur.
d. Eritrosit adalah mikrositik hipokromik dengan poikilositosis, ovalositosis,
dan seringkali bintik-bintik basofil. Sel target mungkin juga ditemukan tapi
biasanya tidak mencolok dan tidak spesifik untuk thalassemia.
Asuhan keperawatan Thalesemia pada anak
Keperawatan Anak II 10
e. MCV rendah, kira-kira 65 fL, dan MCH juga rendah (<26 pg).
Penurunan ringan pada ketahanan hidup eritrosit juga dapat diperlihatkan,
tetapi tanda hemolisis biasanya tidak ada. Kadar besi serum normal atau
meningkat.
4. Thalassemia-β° homozigot (Anemia Cooley, Thalassemia Mayor)
a. bergejala sebagai anemia hemolitik kronis yang progresif selama 6 bulan
kedua kehidupan. Transfusi darah yang reguler diperlukan pada penderita ini
untuk mencegah kelemahan yang amat sangat dan gagal jantung yang
disebabkan oleh anemia. Tanpa transfusi, 80% penderita meninggal pada 5
tahun pertama kehidupan.
b. Pada kasus yang tidak diterapi atau pada penderita yang jarang menerima
transfusi pada waktu anemia berat, terjadi hipertrofi jaringan eritropoetik
disumsum tulang maupun di luar sumsum tulang. Tulang-tulang menjadi tipis
dan fraktur patologis mungkin terjadi. Ekspansi masif sumsum tulang di
wajah dan tengkorak menghasilkan bentuk wajah yang khas.
Asuhan keperawatan Thalesemia pada anak
Keperawatan Anak II 11
Gambar 6. Deformitas tulang pada thalassemia beta mayor
(Facies Cooley)
c. Pucat, hemosiderosis, dan ikterus sama-sama memberi kesan coklat
kekuningan. Limpa dan hati membesar karena hematopoesis ekstrameduler
dan hemosiderosis. Pada penderita yang lebih tua, limpa mungkin sedemikian
besarnya sehingga menimbulkan ketidaknyamanan mekanis dan
hipersplenisme sekunder.
Gambar 7. Splenomegali pada thalassemia
Asuhan keperawatan Thalesemia pada anak
Keperawatan Anak II 12
d. Pertumbuhan terganggu pada anak yang lebih tua; pubertas terlambat atau
tidak terjadi karena kelainan endokrin sekunder. Diabetes mellitus yang
disebabkan oleh siderosis pankreas mungkin terjadi. Komplikasi jantung,
termasuk aritmia dan gagal jantung kongestif kronis yang disebabkan oleh
siderosis miokardium sering merupakan kejadian terminal.
e. Kelainan morfologi eritrosit pada penderita thalassemia-β° homozigot yang
tidak ditransfusi adalah ekstrem. Disamping hipokromia dan mikrositosis berat,
banyak ditemukan poikilosit yang terfragmentasi, aneh (sel bizarre) dan sel target.
Sejumlah besar eritrosit yang berinti ada di darah tepi, terutama setelah splenektomi.
Inklusi intraeritrositik, yang merupakan presipitasi kelebihan rantai α, juga terlihat
pasca splenektomi. Kadar Hb turun secara cepat menjadi < 5 gr/dL kecuali mendapat
transfusi. Kadar serum besi tinggi dengan saturasi kapasitas pengikat besi (iron
binding capacity). Gambaran biokimiawi yang nyata adalah adanya kadar HbF yang
sangat tinggi dalam eritrosit.
3.1.2 Patofisiologi
Thalassemia adalah kelainan herediter dari sintesis Hb akibat dari
gangguan produksi rantai globin. Penurunan produksi dari satu atau lebih
rantai globin tertentu (α,β,γ,δ) akan menghentikan sintesis Hb dan
menghasilkan ketidakseimbangan dengan terjadinya produksi rantai globin
lain yang normal.
Karena dua tipe rantai globin (α dan non-α) berpasangan antara satu sama
lain dengan rasio hampir 1:1 untuk membentuk Hb normal, maka akan terjadi
produksi berlebihan dari rantai globin yang normal dan terjadi akumulasi
Asuhan keperawatan Thalesemia pada anak
Keperawatan Anak II 13
rantai tersebut di dalam sel menyebabkan sel menjadi tidak stabil dan
memudahkan terjadinya destruksi sel. Ketidakseimbangan ini merupakan
suatu tanda khas pada semua bentuk thalassemia. Karena alasan ini, pada
sebagian besar thalassemia kurang sesuai disebut sebagai hemoglobinopati
karena pada tipe-tipe thalassemia tersebut didapatkan rantai globin normal
secara struktural dan juga karena defeknya terbatas pada menurunnya
produksi dari rantai globin tertentu.
Tipe thalassemia biasanya membawa nama dari rantai yang tereduksi.
Reduksi bervariasi dari mulai sedikit penurunan hingga tidak diproduksi sama
sekali (complete absence). Sebagai contoh, apabila rantai β hanya sedikit
diproduksi, tipe thalassemia-nya dinamakan sebagai thalassemia-β+,
sedangkan tipe thalassemia-β° menandakan bahwa pada tipe tersebut rantai β
tidak diproduksi sama sekali. Konsekuensi dari gangguan produksi rantai
globin mengakibatkan berkurangnya deposisi Hb pada sel darah merah
(hipokromatik). Defisiensi Hb menyebabkan sel darah merah menjadi lebih
kecil, yang mengarah ke gambaran klasik thalassemia yaitu anemia
hipokromik mikrositik. Hal ini berlaku hampir pada semua bentuk anemia
yang disebabkan oleh adanya gangguan produksi dari salah satu atau kedua
komponen Hb : heme atau globin. Namun hal ini tidak terjadi pada silent
carrier, karena pada penderita ini jumlah Hb dan indeks sel darah merah
berada dalam batas normal.
Pada tipe trait thalassemia-β yang paling umum, level Hb A2 (δ2/α2)
biasanya meningkat. Hal ini disebabkan oleh meningkatnya penggunaan
Asuhan keperawatan Thalesemia pada anak
Keperawatan Anak II 14
rantai δ oleh rantai α bebas yang eksesif, yang mengakibatkan terjadinya
kekurangan rantai β adekuat untuk dijadikan pasangan. Gen δ, tidak seperti
gen β dan α, diketahui memiliki keterbatasan fisiologis dalam kemampuannya
untuk memproduksi rantai δ yang stabil; dengan berpasangan dengan rantai α,
rantai δ memproduksi Hb A2 (kira-kira 2,5-3% dari total Hb). Sebagian dari
rantai α yang berlebihan digunakan untuk membentuk Hb A2, dimana sisanya
(rantai α) akan terpresipitasi di dalam sel, bereaksi dengan membran sel,
mengintervensi divisi sel normal, dan bertindak sebagai benda asing sehingga
terjadinya destruksi dari sel darah merah. Tingkat toksisitas yang disebabkan
oleh rantai yang berlebihan bervariasi berdasarkan tipe dari rantai itu sendiri
(misalnya toksisitas dari rantai α pada thalassemia-β lebih nyata dibandingkan
toksisitas rantai β pada thalassemia-α).
Dalam bentuk yang berat, seperti thalassemia-β mayor atau anemia
Cooley, berlaku patofisiologi yang sama dimana terdapat adanya substansial
yang berlebihan. Kelebihan rantai α bebas yang signifikan akibat kurangnya
rantai β akan menyebabkan terjadinya pemecahan prekursor sel darah merah
di sumsum tulang (eritropoesis inefektif).
Produksi Rantai Globin
Untuk memahami perubahan genetik pada thalassemia, kita perlu
mengenali dengan baik proses fisiologis dari produksi rantai globin pada
orang sehat atau normal. Suatu unit rantai globin merupakan komponen utama
untuk membentuk Hb : bersama-sama dengan Heme, rantai globin
menghasilkan Hb. Dua pasangan berbeda dari rantai globin akan membentuk
Asuhan keperawatan Thalesemia pada anak
Keperawatan Anak II 15
struktur tetramer dengan Heme sebagai intinya. Semua Hb normal dibentuk
dari dua rantai globin α (atau mirip-α) dan dua rantai globin non-α.
Bermacam-macam tipe Hb terbentuk, tergantung dari tipe rantai globin yang
membentuknya. Masing-masing tipe Hb memiliki karakteristik yang berbeda
dalam mengikat oksigen, biasanya berhubungan dengan kebutuhan oksigen
pada tahap-tahap perkembangan yang berbeda dalam kehidupan manusia.
Pada masa kehidupan embrionik, rantai ζ(rantai mirip-α) berkombinasi
dengan rantai γ membentuk Hb Portland (ζ2γ2) dan dengan rantai ε untuk
membentuk Hb Gower-1 (ζ2ε2).
Selanjutnya, ketika rantai α telah diproduksi, dibentuklah Hb Gower-2,
berpasangan dengan rantai ε (α2ε2). Hb Fetal dibentuk dari α2γ2 dan Hb
dewasa primer (Hb A) dibentuk dari α2β2. Hb fisiologis yang ketiga, Hb A2,
dibentuk dari rantai α2δ2.
Patofisiologi seluler
Kelainan dasar dari semua tipe thalassemia adalah ketidakseimbangan
sintesis rantai globin. Namun, konsekuensi akumulasi dari produksi rantai
globin yang berlebihan berbeda-beda pada tiap tipe thalassemia. Pada
thalassemia-β, rantai α yang berlebihan, tidak mampu membentuk Hb
tetramer, terpresipitasi di dalam prekursor sel darah merah dan, dengan
berbagai cara, menimbulkan hampir semua gejala yang bermanifestasi pada
sindroma thalassemia-β; situasi ini tidak terjadi pada thalassemia-α.
Asuhan keperawatan Thalesemia pada anak
Keperawatan Anak II 16
Rantai globin yang berlebihan pada thalassemia-α adalah rantai γ pada
tahun-tahun pertama kehidupan, dan rantai β pada usia yang lebih dewasa.
Rantai-rantai tipe ini relatif bersifat larut sehingga mampu membentuk
homotetramer yang, meskipun relatif tidak stabil, mampu tetap bertahan
(viable) dan dapat memproduksi molekul Hb seperti Hb Bart (γ4) dan Hb H
(β4). Perbedaan dasar pada dua tipe utama ini mempengaruhi perbedaan besar
pada manifestasi klinis dan tingkat keparahan dari penyakit ini.
Rantai α yang terakumulasi di dalam prekursor sel darah merah bersifat
tidak larut (insoluble), terpresipitasi di dalam sel, berinteraksi dengan
membran sel (mengakibatkan kerusakan yang signifikan), dan mengganggu
divisi sel. Kondisi ini menyebabkan terjadinya destruksi intramedular dari
prekursor sel darah merah. Sebagai tambahan, sel-sel yang bertahan yang
sampai ke sirkulasi darah perifer dengan intracellular inclusion bodies (rantai
yang berlebih) akan mengalami hemolisis; hal ini berarti bahwa baik
hemolisis maupun eritropoesis inefektif menyebabkan anemia pada penderita
dengan thalassemia-β.
Kemampuan sebagian sel darah merah untuk mempertahankan produksi
dari rantai γ, yang mampu untuk berpasangan dengan sebagian rantai α yang
berlebihan untuk membentuk Hb F, adalah suatu hal yang menguntungkan.
Ikatan dengan sebagian rantai berlebih tidak diragukan lagi dapat mengurangi
gejala dari penyakit dan menghasilkan Hb tambahan yang memiliki
kemampuan untuk membawa oksigen.
Asuhan keperawatan Thalesemia pada anak
Keperawatan Anak II 17
Selanjutnya, peningkatan produksi Hb F sebagai respon terhadap anemia
berat, menimbulkan mekanisme lain untuk melindungi sel darah merah pada
penderita dengan thalassemia-β. Peningkatan level Hb F akan meningkatkan
afinitas oksigen, menyebabkan terjadinya hipoksia, dimana, bersama-sama
dengan anemia berat akan menstimulasi produksi dari eritropoetin.
Akibatnya, ekspansi luas dari massa eritroid yang inefektif akan
menyebabkan ekspansi tulang berat dan deformitas. Baik penyerapan besi dan
laju metabolisme akan meningkat, berkontribusi untuk menambah gejala
klinis dan manifestasi laboratorium dari penyakit ini. Sel darah merah
abnormal dalam jumlah besar akan diproses di limpa, yang bersama-sama
dengan adanya hematopoesis sebagai respon dari anemia yang tidak diterapi,
akan menyebabkan splenomegali masif yang akhirnya akan menimbulkan
terjadinya hipersplenisme.
Apabila anemia kronik pada penderita dikoreksi dengan transfusi darah
secara teratur, maka ekspansi luas dari sumsum tulang akibat eritropoesis
inefektif dapat dicegah atau dikembalikan seperti semula. Memberikan
sumber besi tambahan secara teori hanya akan lebih merugikan pasien.
Namun, hal ini bukanlah masalah yang sebenarnya, karena penyerapan besi
diregulasi oleh dua faktor utama : eritropoesis inefektif dan jumlah besi pada
penderita yang bersangkutan. Eritropoesis yang inefektif akan menyebabkan
peningkatan absorpsi besi karena adanya downregulation dari gen HAMP,
yang memproduksi hormon hepar yang dinamakan hepcidin, regulator utama
Asuhan keperawatan Thalesemia pada anak
Keperawatan Anak II 18
pada absorpsi besi di usus dan resirkulasi besi oleh makrofag. Hal ini terjadi
pada penderita dengan thalassemia intermedia.
Dengan pemberian transfusi darah, eritropoesis yang inefektif dapat
diperbaiki, dan terjadi peningkatan jumlah hormon hepcidin; sehingga
penyerapan besi akan berkurang dan makrofag akan mempertahankan kadar
besi.
Pada pasien dengan iron overload (misalnya hemokromatosis), absorpsi
besi menurun akibat meningkatnya jumlah hepsidin. Namun, hal ini tidak
terjadi pada penderita thalassemia-β berat karena diduga faktor plasma
menggantikan mekanisme tersebut dan mencegah terjadinya produksi
hepsidin sehingga absorpsi besi terus berlangsung meskipun penderita dalam
keadaan iron overload.
Efek hepsidin terhadap siklus besi dilakukan melalui kerja hormon lain
bernama ferroportin, yang mentransportasikan besi dari enterosit dan
makrofag menuju plasma dan menghantarkan besi dari plasenta menuju fetus.
Ferroportin diregulasi oleh jumlah penyimpanan besi dan jumlah hepsidin.
Hubungan ini juga menjelaskan mengapa penderita dengan thalassemia-β
yang memiliki jumlah besi yang sama memiliki jumlah ferritin yang berbeda
sesuai dengan apakah mereka mendapat transfusi darah teratur atau tidak.
Sebagai contoh, penderita thalassemia-β intermedia yang tidak mendapatkan
transfusi darah memiliki jumlah ferritin yang lebih rendah dibandngkan
dengan penderita yang mendapatkan transfusi darah secara teratur, meskipun
keduanya memiliki jumlah besi yang sama.
Asuhan keperawatan Thalesemia pada anak
Keperawatan Anak II 19
Kebanyakan besi non-heme pada individu yang sehat berikatan kuat
dengan protein pembawanya, transferrin. Pada keadaan iron overload, seperti
pada thalassemia berat, transferrin tersaturasi, dan besi bebas ditemukan di
plasma. Besi ini cukup berbahaya karena memiliki material untuk
memproduksi hidroksil radikal dan akhirnya akan terakumulasi pada organ-
organ, seperti jantung, kelenjar endokrin, dan hati, mengakibatkan terjadinya
kerusakan pada organ-organ tersebut (organ damage).
Asuhan keperawatan Thalesemia pada anak
Keperawatan Anak II 20
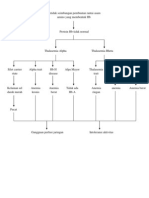
2.1.5 WOC
Primer : genetik, Skunder : Defisiensi asam
idioptaik folat pada kehamilan
Hb post natal terganggu
Gangguan Produksi Rantai
Globin
Penurunan produksi dari 1 atau
lebih rantai globin tertentu Pertumbuhan berlebihan
tulang frontal, zogomatik dan
maxila
Penurunan Sintesis Hb Rantai Beta
Distorsi
tulang muka
Peningkatan Compensatori Sentesa rantai Alfa
Dahi menonjol, mulut tongos,
pertumbuhan gizi tidak teratur
Ketidak seimbangan Formasi hemoglobin
Thalesemia
Penurunan Hb Eritropoesis tidak Pertumbuhan gizi yang
efektif kurang disertai retraksi
tulang rahang
Hipokromatik Penghancuran sel
eritrosit intramedular
Perubahan pada tulang akrena hiperaktivitas
Defisiensi Hb sumsum merah berupa depormitas (pada
Hemolisis kondisi yang tidak atau kurang mendapat
transfusi darah)
Seldarah merah
menjadi kecil Anemia Berat
Suplai nutrisi Anemia
berkurang
Komponen sel darah Anak semakin pucat dan
berkurang Anoreksia mengalami gangguan
pertumbuhan
Berat badan turun
< Hb < O2 Anak semakin
tambak kecil
Hipoksia, sesak Kurangnya selera
Pucat, kelemahan napas makan Penurunan Kemampuan
fisik
Penurunan komponen Ketidakseimbangan Mk : Ketidakseimbangan Mk : Perubahan tumbuh
sel kebutuhan dan suplai nutrisi kurang dari kembang
oksigen kebutuhan
Asuhan keperawatan Thalesemia pada anak
Mk : Perubahan perfusi Mk: Intoleransi
jaringan perifer Aktivitas
Keperawatan Anak II 21
2.1.6 Manifestasi Klinis
Secara klinis talasemia dapat dibagi dalam beberapa tingkatan sesuai
beratnya gejala klinis mayor, intermedia dan minor atau trait (pembawa sifat).
Batas antara tingkatan tersebut sering tidak jelas.
Biasanya bersifat homozygot. Sinonim : Anemia Cooley, Talasemia Beta
Mayor Anemia Mediteranean, Talasemia Homozygot. Gejala klinis berupa
muka mogoloid, pertumbuhan badan kurang sempurna (pendek), pembesaran
hati dan limpa, perubahan pada tulang karena hiperaktivitas sumsum merah
berupa deformitas dan faktor spontan, terutama kasus yang tidak atau kurang
mendapat tranfusi darah. Deformitas tulang disamping mengakibatkan muka
mongoloid, dapat menyebabkan pertumbuhan berlebihan tulang frontal dan
zigomatik serta maksila. Pertumbuhan gizi biasanya buruk. Sering disertai
retraksi tulang rahang. Sinusitis (terutama maksilaris) sering kambuh, akibat
kurang lancarnya drainase pertumbuhan intelektual dan berbicara biasanya
tidak terganggu. IQ kurang baik apabila tidak mendapat tranfusi darah secara
teratur dan cukup menaikkan kadar Hb.
Anemia biasanya berat dan biasanya mulai muncul gejalanya pada usia
beberapa bulan serta menjadi jelas pada usia 2 tahun. Ikterus jarang terjadi
dan bila ada biasanya ringan. Talasemia -bo homozygot pada umumnya
memerlukan tranfusi secara reguler, tetapi ada kalanya berlangsung ringan
dan memberikan gambaran klinis seperti talasemia intermedia. Talasemia beta
diantara orang negro (talasemia beta 2) pada umumnya berlangsung ringan.
Asuhan keperawatan Thalesemia pada anak
Keperawatan Anak II 22
Pada talasemia intermedia dan minor sesuai dengan arti katanya
didapatkan variasi luas mengenai jenis gejala klinis. Talasemia intermedia
fenotipik adalah talasemia mayor tanpa adanya kerusakan gen. Keadaan
klinisnya lebih baik dan gejala lebih ringan daripada talasemia mayor. Pada
talasemia intermedia umumnya tidak ada splenomegali. Anemia ringan, bila
ada disebabkan oleh masa hidup eritrosit yang memendek.
Pada talasemia trait umumnya tidak dijumpai gejala klinis yang khas.
Hanya di dapat kelainan pada eritrosit dan atau hanya sebagian dari gejala
yang didapat pada kasus homozygot.
Gambaran klinis penyakit talasemia beta Hb E menyerupai talasemia
mayor Hb dalam hal ini terdiri dari HbE, HbF dan apabila ada Hb A1 dalam
jumlah yang sedikit.
Talesemia mayor mulai menunjukkan gejala anemia pada masa bayi
(kadang – kadang pada umur 3 bulan) pada waktu sintesis rantai -b
menggantikan sintesis rantai - l. Anak semakin pucat dan mengalami
gangguan pertumbuhan sehingga makin nyata tampak kecil, fragil. Lama –
lama perut membuncit karena splenomegali. Karena itu setiap anak dengan
pucat (terutama bila anemia berat), fragil, mungkin juga ditemukan PEM I
maka dia harus dicurigai menderita talasemia, mengingat Indonesia adalah
daerah sindrom talasemia. Pada pengamatan lebih dekat tampak muka
mongoloid dengan hipertolerisme, nasal bridge pesek; pada anak yang agak
besar mulut tonggos (rodent like mouth) akibat maksila yang lebih menonjol,
bibir atas agak terangkat. Splenomegali makin nyata dengan makin
Asuhan keperawatan Thalesemia pada anak
Keperawatan Anak II 23
bertambahnya umur. Hepatomegali umumnya ada, pasca splenektomi
hepatomegali selalu ada dan progresif. Limfadenopati jarang terjadi.
Pada masa remaja terjadi keterlambatan menarche dan pertumbuhan alat
kelamin sekunder, keterlambatan fungsi reproduksi. Dapat pula terjadi fraktur
patologik, ulkus kronik ditungkai bawah seperti pada anemia hemolitik kronik
yang lain sebagai akibat dari ekspansi eritropoesis. Terjadi distorsi tulang –
tulang muka sehingga dahi menonjol, mulut tonggos, pertumbuhan gigi tidak
teratur.
Hemosiderosis makin nyata pada dekade kedua kehidupan terutama pada
penderita yang sering mendapat tranfusi (sampai > 100 kali) dan tidak
mendapat iron chelating agent untuk mengeluarkan timbunan besi tubuh.
Pada Rontgen tulang kepala tampak gambaran “hair on end” korteks tipis
bahkan tak tampak, diploe tampak seperti garis – garis tegak lurus pada
lengkung tengkorak seperti gambaran singkat.
2.1.7 Pemeriksaan Penunjang
1. Darah tepi
- Hb rendah dapat sampai 2 atau 3 gr%
- Gambaran morfologi eritrosit : mikrositik hipokromik, sel target,
anisositosis berat dengan makrovaloositosis, mikrosferosit, polikromasi,
basophilic stippling, benda Howell – jolly, poikilositosis dan sel target.
Gambaran ini lebih kurang khas.
- Normoblas di daerah tepi terutama jenis asidofil (perhatikan normoblas
adalah sel darah merah yang masih berinti sehingga ikut terhitung pada
Asuhan keperawatan Thalesemia pada anak
Keperawatan Anak II 24
perhitungan lukosit dengan bilik hitung adalah AL lebih tinggi dari
pada sebenarnya)
- Retikulosit meninggi
2. Susunan Tulang (tidak menentukan diagnosis)
- Hiperplasi sistem eritropoesis dengan normoblas terbanyak dari jenis
asidofil.
- Granula Fe (dengan pengecatan prussian Blue) meningkat.
3. Pemeriksaan Khusus
- HbF meninggi : 20% - 90% Hb total (alkali denaturasi)
- Elektroforesis Hb untuk menunjukkan hemoglobinopati yang lain
maupun mengukur kadar HbF.
- Pemeriksaan pedigree untuk memastikan diagnosis : kedua orang tua
pasien telasemia mayor merupakan trait (carier) dengan Hb A2 meninggi
(> 3,5 dari Hb total)
4. Pemeriksaan Lain
- Foto Ro tulang kepala menunjukkan gambaran hair on end kortex
menipis, diploe melebar dengan traberkula tegak lurus pada kortex.
- Foto tulang pipih dan ujung tulang panjang menunjukkan perluasan
sumsum tulang ® trabekula tampak jelas.
- Fragilitas eritrosit terhadap larutan NaCl menurun
- Bukti pasti fenotif talasemia adalah ketidakseimbangan produksi rantai
polipeptida globin (diagnosis molekuler)
Asuhan keperawatan Thalesemia pada anak
Keperawatan Anak II 25
2.1.8 Penatalaksanaan
Medikamentosa
Pemberian iron chelating agent (desferoxamine): diberikan
setelah kadar feritin serum sudah mencapai 1000 g/l atau
saturasi transferin lebih 50%, atau sekitar 10-20 kali transfusi
darah.
Desferoxamine, dosis 25-50 mg/kg berat badan/hari subkutan
melalui pompa infus dalam waktu 8-12 jam dengan minimal selama
5 hari berturut setiap selesai transfusi darah.
Vitamin C 100-250 mg/hari selama pemberian kelasi besi, untuk
meningkatkan efek kelasi besi.
Asam folat 2-5 mg/hari untuk memenuhi kebutuhan yang meningkat.
Vitamin E 200-400 IU setiap hari sebagai antioksidan dapat
memperpanjang umur sel darah merah.
II. Bedah
Splenektomi, dengan indikasi:
limpa yang terlalu besar, sehingga membatasi gerak penderita,
menimbulkan peningkatan tekanan intraabdominal dan bahaya
terjadinya ruptur
hipersplenisme ditandai dengan peningkatan kebutuhan transfusi
darah atau kebutuhan suspensi eritrosit (PRC) melebihi 250 ml/kg
berat badan dalam satu tahun.
Asuhan keperawatan Thalesemia pada anak
Keperawatan Anak II 26
III. Suportif
Transfusi darah :
Hb penderita dipertahankan antara 8 g/dl sampai 9,5 g/dl. Dengan kedaan
ini akan memberikan supresi sumsum tualang yang adekuat, menurunkan
tingkat akumulasi besi, dan dapat mempertahankan pertumbuhan dan
perkembangan penderita. Pemberian darah dalam bentuk PRC (packed
red cell), 3 ml/kg BB untuk setiap kenaikan Hb 1 g/dl.
IV. Lain-lain (rujukan subspesialis, rujukan spesialisasi lainnya dll)
Tumbuh kembang, kardiologi, Gizi, endokrinologi, radiologi, Gigi
2.1.9 Komplikasi
Akibat anemia yang berat dan lama, sering terjadi gagal jantung. Transfusi
darah yang berulang-ulang dan proses hemolisis menyebabkan kadar besi
dalam darah tinggi, sehingga ditimbun dalam berbagai jaringan tubuh seperti
hepar, limpa, ku.lit, jantung dan lainnya. Hal ini dapat mengakibatkan
gangguan fungsi alat tersebut. Limpa yang besar mudah rupture akibat trauma
yang ringan. Kadang-kadang thalasemia disertai oleh tanda hipersplenisme
seperti leukopenia dan trombopenia.
Kematian terutama disebabkan oleh infeksi dan gagal jantung.
Kelebihan Fe (khususnya pada pemberian transfusi)
Komplikasi pada jantung, contoh constrictive pericarditis to heart failure
and arrhythmias.
Komplikasi pada hati, contoh hepatomegali sampai cirrhosis.
Asuhan keperawatan Thalesemia pada anak
Keperawatan Anak II 27
Komplikasi jangka panjang, contoh HCV.
Komplikasi hematologic, contoh VTE.
Komplikasi pada endokrin, seperti endokrinopati, DM.
Gagal tumbuh karena diversi dari sumber kalori untuk eritropoesis.
Fertil, seperti terjadi hypogonadotrophic hypogonadism dan gangguan
kehamilan.
2.2 Asuhan Keperawatan Teoritis
2.2.1 Pengkajian
A. Asal Keturunan / Kewarganegaraan
• Thalasemia banyak dijumpai pada bangsa di sekitar laut Tengah
(Mediteranial) seperti Turki, Yunani, dll. Di Indonesia sendiri, thalasemia
cukup banyak dijumpai pada anak, bahkan merupakan penyakit darah
yang paling banyak diderita.
B. Umur
• Pada penderita thalasemia mayor yang gejala klinisnya jelas, gejala telah
terlihat sejak anak berumur kurang dari 1 tahun, sedangkan pada
thalasemia minor biasanya anak akan dibawa ke RS setelah usia 4 tahun.
C. Riwayat Kesehatan Anak
• Anak cenderung mudah terkena infeksi saluran pernapasan atas atau
infeksi lainnya. Ini dikarenakan rendahnya Hb yang berfungsi sebagai
alat transport.
D. Pertumbuhan dan Perkembangan
Asuhan keperawatan Thalesemia pada anak
Keperawatan Anak II 28
• Seirng didapatkan data adanya kecenderungan gangguan terhadap
tumbang sejak masih bayi. Terutama untuk thalasemia mayor,
pertumbuhan fisik anak, adalah kecil untuk umurnya dan adanya
keterlambatan dalam kematangan seksual, seperti tidak ada pertumbuhan
ramput pupis dan ketiak, kecerdasan anak juga mengalami penurunan.
Namun pada jenis thalasemia minor, sering terlihat pertumbuhan dan
perkembangan anak normal.
E. Pola Makan
• Terjadi anoreksia sehingga anak sering susah makan, sehingga BB rendah
dan tidak sesuai usia.
F. Pola Aktivitas
• Anak terlihat lemah dan tidak selincah anak seusianya. Anak lebih
banyak tidur/istirahat karena anak mudah lelah.
G. Riwayat Kesehatan Keluarga
• Thalasemia merupakan penyakit kongenital, jadi perlu diperiksa apakah
orang tua juga mempunyai gen thalasemia. Jika iya, maka anak beresiko
terkena talasemia mayor.
H. Riwayat Ibu Saat Hamil (Ante natal Core – ANC)
• Selama masa kehamilan, hendaknya perlu dikaji secara mendalam
adanya faktor resiko talasemia. Apabila diduga ada faktor resiko, maka
ibu perlu diberitahukan resiko yang mungkin sering dialami oleh anak
setelah lahir.
I. Data Keadaan Fisik Anak Thalasemia
Asuhan keperawatan Thalesemia pada anak
Keperawatan Anak II 29
• KU = lemah dan kurang bergairah, tidak selincah anak lain yang seusia.
• Kepala dan bentuk muka. Anak yang belum mendapatkan pengobatan
mempunyai bentuk khas, yaitu kepala membesar dan muka mongoloid
(hidung pesek tanpa pangkal hidung), jarak mata lebar, tulang dahi
terlihat lebar.
• Mata dan konjungtiva pucat dan kekuningan
• Mulut dan bibir terlihat kehitaman
• Dada, Pada inspeksi terlihat dada kiri menonjol karena adanya
pembesaran jantung dan disebabkan oleh anemia kronik.
• Perut, Terlihat pucat, dipalpasi ada pembesaran limpa dan hati
(hepatospek nomegali).
• Pertumbuhan fisiknya lebih kecil daripada normal sesuai usia, BB di
bawah normal
• Pertumbuhan organ seks sekunder untuk anak pada usia pubertas tidak
tercapai dengan baik. Misal tidak tumbuh rambut ketiak, pubis ataupun
kumis bahkan mungkin anak tidak dapat mencapai tapa odolense karena
adanya anemia kronik.
• Kulit, Warna kulit pucat kekuningan, jika anak telah sering mendapat
transfusi warna kulit akan menjadi kelabu seperti besi. Hal ini terjadi
karena adanya penumpukan zat besi dalam jaringan kulit (hemosiderosis).
2.2.2 Diagnosa Keperawatan Yang Mungkin muncul
1. Perubahan perfusi jaringan b.d berkurangnya komponen seluler yang
penting untuk menghantarkan Oksigen/zat nutrisi ke sel.
Asuhan keperawatan Thalesemia pada anak
Keperawatan Anak II 30
2. Intoleransi aktivitas b.d ketidakseimbangan kebutuhan pemakaian dan
suplai oksigen.
3. Ketidakseimbangan nutrisi kurang dari kebutuhan b.d kurangnya selera
makan.
4. Koping keluarga tidak efektif b.d dampak penyakit anak terhadap fungsi
keluarga.
5. Resiko terjadi kerusakan integritas kulit berhubungan dengan perubahan
sirkulasi dan neurologis.
6. Resiko infeksi berhubungan dengan pertahanan sekunder tak adekuat:
penurunan Hb, leukopeni atau penurunan granulosit.
7. Perubahan tumbuh kembang berhubungan dengan penurunan
kemampuan fisik yang disebabkan oleh kelainan hematology dan efek
penyakit dan terapi.
Asuhan keperawatan Thalesemia pada anak
Keperawatan Anak II 31
2.2.3 Rencana Asuhan Keperawatan
DIAGNOSA TUJUAN DAN KRITERIA
NO INTERVENSI RASIONAL
KEPERAWATAN HASIL
1 Perubahan perfusi Setelah dilakukan tindakan - Monitor - Perubahan tanda
jaringan b.d keperawatan selama 5x 24 jam TTV,pengisian vital,warna kulit dan
berkurangnya perfusi jaringan klien adekuat kapiler,warna kulit dan membran mukosa
komponen seluler dengan criteria : membaran mukosa menunjukkan tanda
yang penting untuk - Membran mukosa merah muda perfusi jaringan
menghantarkan - Conjunctiva tidak anemis - Tinggikan posisi - Meningkatkan ekspansi
oksigen/zat nutrisi - Akral hangat kepala tempat tidur paru dan memaksimalkan
- TTV dalam batas normal oksigen untuk kebutuhan
seluler
- Periksa adanya - Iskemia seluler
keluhan nyeri mempengaruhi
jar.miokardial
- Catat keluhan rasa - Vasokontriksi ke organ
dingin vital menurunkan
- Pertahankan suhu sirkulasi perifer
lingkungan dan tubuh
hangat - Memaksimalkan transfer
- Beri oksigen sesuai oksigen ke jaringan
kebutuhan - Memantau kadar
oksigenasi
- Kolaborasi dalam
Setelah diberikan tindakan pemeiksaan lab :
2 keperawatan selama 3 x 24 jam HB,HMT,SDM. - Mempengaruhi pilihan
Intoleransi aktivitas klien toleran terhadap aktivitas intervensi
b.d dengan criteria : - Kaji kemampuan anak
Asuhan keperawatan Thalesemia pada anak
Keperawatan Anak II 32
ketidakseimbangan - Kebutuhan ADL dalm melakukan - Manifestasi
kebutuhan pemakaian terpenuhi tanpa rasa aktivitas/memenuhi kardiopulmonal dari
dan suplai oksigen pusing,sesak ADL upaya jantung dan paru
- Monitor TTV,respon untuk membawa jml
fisiologis oksigen adekuat ke jar.
selama,setelah - Rangsangan/stress
melakukan aktivitas kardiopulmonal
berlebihan dpt
- Beri informasi pada menimbulkan
anak/klg untuk dekompensasi/kegagalan
berhenti melakukan - Membantu dan memberi
aktivitas jika terjadi dukungan
peningkatan TTV
atau pusing - Memperthanan tingkat
Setelah diberikan tindakan - Beri bantuan dalam energi dan meningkatkan
keperawatan selama 3 x 24 jam beraktivitas/ambulasi regangan pada system
nutrisi klien terpenuhi dengan ila perlu jantung dan pernafasan.
3 criteria - Perioritaskan jadwal
- BB stabil/meningkat askep untuk - Mengidentifikasi
Ketidakseimbangan - Nilai laboratorium Dbn meningkatkan defisiensi,merencanakan
nutrisi kurang dari - Melaporkan nafsu makan istirahat intervensi
kebutuhan b.d meningkat - Mengawasi masukan
kurangnya selera - Menghabiskan porsi makan yang kalori atau kualitas
makan disediakan. - Kaji riwayat nutrisi kekurangan konsumsi
dan makanan yg makanan
disukai - mengawasi penurunan
BB atau efektivitas
- Observasi dan catat intervensi nutrisi
masukan makanan - Makan dpt menurunkan
kelemahan dan
Asuhan keperawatan Thalesemia pada anak
Keperawatan Anak II 33
meningkatkan
- Timbang Berat badan pemasukan juga
setiap hari mencegah distensi gaster
- Membantu membuat
Setelah diberikan tindakan - Beri makanan sedikit rencana diet
keperawatan selama 3 x 24 jam tapi sering dan atau - Menigkatkan masukan
keluarga dapat mengatasi dan makan diantara waktu protein dan kalori
4 mengendalikan stress yang terjadi makan
pada keluarga dengan criteria : - Keluarga paham dengan
kondisi anak dan dapat
- Keluarga menerima kondisi
Koping Keluarga tidak - Konsul ahli gizi menerima sesuai keadaan
anaknya
efektif b.d dampak - Orang terdeklat
- Menunjukkan tingkah laku koping
penyaklit anak - Beri obat/suplemen memerlukan dukungan
yang positip
terhadap fungsi vitamin sesuai order yg terus menerus dg
keluarga berbagai masalah yg
- Jelaskan kondisi anak dihadapi akan
sesuai realita dan beri meningkatkan dlm
dukungan pada mengatasi penyakit untuk
keluarga memudahkan proses
- Berikan adaptasi
waktu/dengarkan hal- - Dukungan keluarga thd
hal yang mejadi anak dapat meningktkan
keluhan keluarga harapan anak
- Tingkah laku yang
terhalang,tuntutan
perawatan tinggi dan
seterusnya dapat
- Memberikan menimbulkan klg
dukungan kepada menarik diri dri
Asuhan keperawatan Thalesemia pada anak
Keperawatan Anak II 34
keluarga untuk pergaulan social.
mengembangkan
harapan realistis thd
anak
- Bantu keluarga untuk
memahami betapa
pentingnya
mempertahankan
fungsi psikososial
Asuhan keperawatan Thalesemia pada anak
Keperawatan Anak II 35
BAB IV
PENUTUP
4.1 Kesimpulan
Thalassemia adalah gangguan pembuatan hemoglobin yang diturunkan.
Thalassemia ditemukan tersebar di seluruh ras di Mediterania, Timur Tengah, India
sampai Asia Tenggara. Thalassemia memiliki dua tipe utama berdasarkan rantai
globin yang hilang pada hemoglobin individu yaitu Thalassemia-α dan thalassemia-
β, yang nantinya akan dibagi lagi menjadi beberapa subtipe berdasarkan derajat
mutasi (secara genetik) ataupun berat ringannya gejala. Thalassemia diturunkan
berdasarkan hukum Mendel, resesif atau ko-dominan. Heterozigot biasanya tanpa
gejala, sedangkan homozigot atau gabungan heterozigot gejalanya lebih berat dari
thalassemia α dan β. Terapi thalassemia antara lain adalah terapi transfusi, terapi
pengikat besi (khelasi), splenektomi, dan transplantasi sumsum tulang. Masing-
masing terapi memiliki kriteria dan efek samping tertentu sehingga perlu
dipertimbangkan secara seksama. Konseling mengenai thalassemia sangat diperlukan
untuk skrining dan pemahaman terhadap penderita. Sampai saat ini, penderita
thalassemia yang berat biasanya tidak dapat bertahan hingga mencapai usia dewasa
normal meskipun kemungkinan ini tidak tertutup sama sekali.
Asuhan keperawatan Thalesemia pada anak
Keperawatan Anak II 36
DAFTAR PUSTAKA
Behrman, Kliegman, Arvin. Nelson : Ilmu Kesehatan Anak Volume 2. Edisi ke-15.
Jakarta : EGC ; 1996
Doenges, Marillyn E. 1999.Rencana Asuhan Keperawatan.
Ngastiyah.1997.Perawatan Anak Sakit. Penerbit Buku Kedokteran EGC.Jakarta
http://askep-askeb.cz.cc/2010/08/asuhan-keperawatan-talasemia.html
http://www.docstoc.com/docs/28719352/LP-TALASEMIA
Kosasih, E.N. 2001. Buku Ajar Ilmu Penyakit Dalam. Jilid II. Edisi ketiga. Jakarta:
Balai Penerbit FKUI
Mansjoer, Kapita selekta kedokteran Ed 3, jilid 2 Media Aesculapius Jakarta : 1999
Ikatan Dokter Anak Indonesia. Buku Ajar Hematologi-Onkologi Anak. 2005. Jakarta:
Badan Penerbit IDAI
Permono B, Sutaryo, dkk. Buku Ajar Hemotologi-Onkologi Anak Cetakan Kedua.
Jakarta : Ikatan Dokter Anak Indonesia ; 2006
Wahidiyat I, Thalassemia dan Permasalahannya di Indonesia. Naskah lengkap Kongres
Nasional Ilmu Kesehatan Anak (KONIKA) Jakarta, 1999 : 293-6
Asuhan keperawatan Thalesemia pada anak
Keperawatan Anak II 37
Defesiensi
Hb
KATA PENGANTAR
Puji dan syukur penulis panjatkan pada ALLAH SWT yang telah memberikan
rahmat dan karunia-Nya sehingga penulis dapat menyelesaikan tugas mata kuliah
Keperawatan anak II dengan judul “ Asuhan Keperawatn Thalesemia pada Aanak “
tepat waktu.
Makalah ini disampaikan untuk memenuhi kelengkapan syarat penilaian mata
kuliah Keperawatan anak II. Adapun kata-kata yang terdapat dalam makalah ini penulis
ambil dari sumber-sumber referensi yang berkaitan dengan judul yang telah ditentukan.
Berkenaan dengan makalah ini penulis menyampaikan ucapan terimakasih kepada Ns.
Hanifah, S.Kep sebagai dosen pengajar mata kuliah Keperawatan Anak II yang telah
memberikan ilmu mengenai Keperawatan Anak II kepada penulis sehingga menambah
wawasan dan pengetahuan bagi penulis. Tak lupa penulis juga menyampaikan
terimakasih kepada semua pihak yang telah membantu dalam menyelesaikan makalah
ini.
Penulis mengharapkan semoga dengan makalah ini dapat menambah lebih
banyak wawasan dan pengetahuan. Diharapkan juga makalah ini dapat menunjang
kelengkapan syarat penilaian mata kuliah psikiatri. Disamping itu, penulis mohon maaf
apabila terdapat kekeliruan pada makalah ini.
Bengkulu, Oktober 2010
Penulis
Asuhan keperawatan Thalesemia pada anak
Keperawatan Anak II 38
DAFTAR ISI
HALAMAN JUDUL................................................................................................
KATA PENGANTAR............................................................................................ .
DAFTAR ISI.............................................................................................................
BAB I PENDAHULUAN
A. Latar Belakang......................................................................................... 1
B. Tujuan....................................................................................................... 2
C. Manfaat..................................................................................................... 2
BAB II LANDASAN TEORI
1. Konsep Teori Thalesemia
A. Definisi..................................................................................................... 3
B. Etiologi..................................................................................................... 4
C. Klasifikasi Thalesemia............................................................................. 5
D. Patofisiologi............................................................................................. 13
E. WOC........................................................................................................ 22
F Manifestasi Klinis..................................................................................... 23
G. Pemeriksaan Penunjang........................................................................... 25
H. Penatalaksanaan....................................................................................... 27
I. Komplikasi............................................................................................... 28
2. Asuhan Keperawatan teoritis
A. Pengkajian................................................................................................ 29
B. Diagnosa Keperawatan yang mungkin muncul....................................... 31
C. Rencana Asuhan Keperawatan................................................................. 33
BAB III Tinjauan Kasus........................................................................................... 36
BAB IV Penutup
A. Kesimpulan.............................................................................................. 46
DAFTAR PUSTAKA............................................................................................... 47
Asuhan keperawatan Thalesemia pada anak
Keperawatan Anak II 39
MAKALAH
Keperawatan Anak II
“Asuhan Keperawatan Thalesemia Pada Anak”
Dosen
Ns. Hanifah, S.Kep
Dissun Oleh :
MEILTHA SURYAWAN
(0726010046)
TIA UTARI (0726010042)
REKA NOPRIANA (0726010022)
ANI TRISNA WATI (0726010096)
RIKI ROSADI (0726010088)
MINARNA (0726010072)
EVA SEPTIANI (0626010054)
IRA NOVITA SARI (0626010047)
SEKOLAH TINGGI ILMU KESEHATAN
TRI MANDIRI SAKTI
BENGKULU
Asuhan keperawatan Thalesemia pada anak
Keperawatan Anak II 40
2010
Asuhan keperawatan Thalesemia pada anak
You might also like
- Laporan Pendahuluan ThalasemiaDocument35 pagesLaporan Pendahuluan ThalasemiaDiah Dwi Meidayanti100% (1)
- Anak ThalasemiaDocument27 pagesAnak ThalasemiaYulia PanmaNo ratings yet
- LP ThalasemiaDocument17 pagesLP ThalasemiaChandra100% (1)
- LAPORAN PENDAHULUAN ThalasemiaDocument9 pagesLAPORAN PENDAHULUAN ThalasemiaOchenRmpVNo ratings yet
- Satuan Acara Penyuluhan ThalasemiaDocument10 pagesSatuan Acara Penyuluhan ThalasemiasantistianaNo ratings yet
- Pathway ThalasemiaDocument1 pagePathway ThalasemiaRony NKcom0% (1)
- Asuhan Keperawatan Pada Anak Dengan ThalassemiaDocument30 pagesAsuhan Keperawatan Pada Anak Dengan ThalassemiaFirman SyahNo ratings yet
- Askep Thalasemia Pada Anak Kel 2Document13 pagesAskep Thalasemia Pada Anak Kel 2majid nugrahaNo ratings yet
- Asuhan Keperawatan Pada Anak AnemiaDocument23 pagesAsuhan Keperawatan Pada Anak Anemianungki nurseptiNo ratings yet
- Intervensi Keperawatan Pasien KritisDocument15 pagesIntervensi Keperawatan Pasien KritisEpull EmpungNo ratings yet
- Askep Anemia Sel SabitDocument17 pagesAskep Anemia Sel SabitWahyuni JayantiNo ratings yet
- Asuhan Keperawatan Aplikasi NANDA SLE Nic NocDocument22 pagesAsuhan Keperawatan Aplikasi NANDA SLE Nic NocSusmianah0% (1)
- Askep Thalasemia Pada AnakDocument18 pagesAskep Thalasemia Pada AnakRezki wahyuniNo ratings yet
- Askep Thalasemia Pada AnakDocument28 pagesAskep Thalasemia Pada AnaksugarviciNo ratings yet
- (CI) 4. LP Thalasemia (Poli Anak)Document4 pages(CI) 4. LP Thalasemia (Poli Anak)Ella Effendi22No ratings yet
- LP ThalasemiaDocument12 pagesLP ThalasemiaSUGENG WINOTO100% (3)
- Bab Ii DDSTDocument23 pagesBab Ii DDSTanekefadilaNo ratings yet
- Askep IspaDocument11 pagesAskep Ispawidchia widya tanNo ratings yet
- Askep AnemiaDocument32 pagesAskep AnemiaartantiNo ratings yet
- HaemofiliaDocument22 pagesHaemofiliaabdul muisNo ratings yet
- LP ThalasemiaDocument2 pagesLP Thalasemiaendah sevianaNo ratings yet
- Askep Keluarga Dewi BaruDocument21 pagesAskep Keluarga Dewi Barusiti mardewiNo ratings yet
- ThalasemiaDocument31 pagesThalasemiagery tampiNo ratings yet
- Asuhan Keperawatan Pada Pasien StrokeDocument14 pagesAsuhan Keperawatan Pada Pasien StrokeNur Mazila100% (1)
- Asuhan Keperawan Medikal Bedah Penyakit Gagal GinjalDocument21 pagesAsuhan Keperawan Medikal Bedah Penyakit Gagal GinjalKRISTIAN ABRAHAM MAILOANo ratings yet
- Askep Thalasemia Pada AnakDocument11 pagesAskep Thalasemia Pada AnakYogi Iqbal FirdausNo ratings yet
- Asuhan Keperawatan Ibu Hamil Dengan Diabetes Melitus GestasionalDocument12 pagesAsuhan Keperawatan Ibu Hamil Dengan Diabetes Melitus GestasionalAndre HamzahNo ratings yet
- Aml LP OkDocument29 pagesAml LP OkAnitha HarusTetap TegarhadapiSemuaNo ratings yet
- LP Pneumonia BeratDocument18 pagesLP Pneumonia BeratLevhy Cwoq NyebeliinNo ratings yet
- LeukemiaDocument32 pagesLeukemiadwi chandraNo ratings yet
- Laporan Pendahuluan ThalasemiaDocument41 pagesLaporan Pendahuluan ThalasemiaNur Oktavia RhosaniNo ratings yet
- Askep Bumil Dengan Anemia FebrilDocument3 pagesAskep Bumil Dengan Anemia FebrilFebrilianti Kusuma WardhaniNo ratings yet
- LP DHF FixDocument29 pagesLP DHF FixadeNo ratings yet
- Askep TalasemiaDocument31 pagesAskep TalasemiaMega Amelia100% (1)
- Respiratory Distress SyndromeDocument22 pagesRespiratory Distress SyndromeDie'an WhkleeiyYiNo ratings yet
- LP LeukemiaDocument14 pagesLP LeukemiaintanayuNo ratings yet
- ASKEP DHFDocument14 pagesASKEP DHFGedeSantosa100% (1)
- Askep RetinoblastomaDocument12 pagesAskep RetinoblastomaOverlinda limbongNo ratings yet
- Woc EklampsiaDocument6 pagesWoc Eklampsiadian_darmalini96No ratings yet
- Rizky Gusti Saleh (A2r17029) Kasus AnemiaDocument48 pagesRizky Gusti Saleh (A2r17029) Kasus Anemiairma soviya aNo ratings yet
- LP Leukemia.Document11 pagesLP Leukemia.Aziz Nur FathoniNo ratings yet
- Nuur Kumala Sari Dewi - LP-LK - Dehidrasi SedangDocument22 pagesNuur Kumala Sari Dewi - LP-LK - Dehidrasi SedangAni ArdiantiNo ratings yet
- LP Hiv KMB 2Document25 pagesLP Hiv KMB 2ErinadhrdnNo ratings yet
- Askep ItpDocument13 pagesAskep ItpSilviaoktaherlianiNo ratings yet
- Askep Anemia AnakDocument12 pagesAskep Anemia AnakPradhevi Angelita WijayaNo ratings yet
- Perawatan Anak Sakit Di RumahDocument2 pagesPerawatan Anak Sakit Di RumahRamadhan GhaffarNo ratings yet
- Laporan Pendahuluan CopdDocument23 pagesLaporan Pendahuluan CopdirwanNo ratings yet
- Thalasemia Pada AnakDocument21 pagesThalasemia Pada AnakLalu Fathul AzizNo ratings yet
- Komunikasi Terapeutik Pada LansiaDocument15 pagesKomunikasi Terapeutik Pada LansiaNovena MoningkaNo ratings yet
- Bab 3 BK FixxxxDocument89 pagesBab 3 BK FixxxxApriliaNo ratings yet
- Pathway DHF Dan KDMDocument2 pagesPathway DHF Dan KDMssandralianiNo ratings yet
- ToksoplasmosisDocument9 pagesToksoplasmosisSanty MirdaNo ratings yet
- Askep PDA Pada AnakDocument18 pagesAskep PDA Pada AnakPadila PadilaNo ratings yet
- Diagnosis Terkini Dan Tatalaksana DHFDocument75 pagesDiagnosis Terkini Dan Tatalaksana DHFDwi Prasetyo Nugroho100% (1)
- Satuan Acara Penyuluhan ThalasemiaDocument5 pagesSatuan Acara Penyuluhan ThalasemiaAuristi MulfitriNo ratings yet
- LP Anak Thalasemia Ruslandi - SKEPDocument22 pagesLP Anak Thalasemia Ruslandi - SKEPSalman FirmansyahNo ratings yet
- LP Thalasemia KosiDocument15 pagesLP Thalasemia KosiLiztNo ratings yet
- Makalah Keperawatan Anak Bu Antarini (Itp & Trombositopenia)Document45 pagesMakalah Keperawatan Anak Bu Antarini (Itp & Trombositopenia)Sally Violetta TamaraNo ratings yet
- Tugas ThalasemiaDocument13 pagesTugas ThalasemiamutiaraNo ratings yet
- Laporan Pendahuluan ThalasemiaDocument20 pagesLaporan Pendahuluan ThalasemiaNurulmutmainnahNo ratings yet
- Manajeman RSSMDocument54 pagesManajeman RSSMQ Chayanx KhamoeNo ratings yet
- WOC ThalesemiaDocument2 pagesWOC ThalesemiaQ Chayanx KhamoeNo ratings yet
- Perubahan OrganisasiDocument10 pagesPerubahan Organisasiiyandri tiluk wahyonoNo ratings yet
- Resep Nasi Goreng IstimewaDocument4 pagesResep Nasi Goreng IstimewaQ Chayanx KhamoeNo ratings yet