Professional Documents
Culture Documents
Fond Amen Ti Di Termodinamica Dell Ingegneria Chimica
Uploaded by
Marco Lawrence NewkirkCopyright
Available Formats
Share this document
Did you find this document useful?
Is this content inappropriate?
Report this DocumentCopyright:
Available Formats
Fond Amen Ti Di Termodinamica Dell Ingegneria Chimica
Uploaded by
Marco Lawrence NewkirkCopyright:
Available Formats
Fondamenti di
Termodinamica
dellIngegneria Chimica
Renato Rota
sempre molto difficile
ringraziare tutti coloro che hanno
contribuito alla stesura di un libro.
Fra tutti per giusto ricordare
Sylvie e Michele, che
apparentemente non centrano
nulla. Oltre, naturalmente, alla
bistopa.
Complicare facile, semplificare
difficile.
Bruno Munari
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
I
Sommario
1 Definizioni e concetti propedeutici ................................................................................1
1.1 Leggi della termodinamica.....................................................................................1
1.2 Sistemi termodinamici............................................................................................2
1.3 Propriet termodinamiche e unit di misura...........................................................3
1.3.1 Sistema Internazionale ....................................................................... 5
1.3.2 Analisi dimensionale ........................................................................ 11
1.3.3 Sistema Anglosassone ...................................................................... 11
1.3.4 Altre unit di misura......................................................................... 12
1.4 Trasformazioni spontanee e condizioni di equilibrio ...........................................13
2 Bilanci in sistemi monocomponente ............................................................................19
2.1 Formulazione generale di un bilancio ..................................................................19
2.2 Bilancio di materia ...............................................................................................20
2.2.1 Condizioni stazionarie...................................................................... 21
2.2.2 Applicazione..................................................................................... 21
2.3 Bilancio di energia ...............................................................................................22
2.3.1 Condizioni stazionarie...................................................................... 25
2.3.2 Primo principio della termodinamica ............................................... 26
2.3.2.1 Trasformazioni semplici...........................................................................28
2.3.2.2 Stati di riferimento....................................................................................31
2.3.3 Applicazione..................................................................................... 31
2.4 Bilancio di entropia ..............................................................................................33
2.4.1 Condizioni stazionarie...................................................................... 33
2.4.2 Secondo principio della termodinamica ........................................... 34
2.4.3 Combinazione del primo e del secondo principio ............................ 37
2.5 Gas perfetto ..........................................................................................................38
2.5.1 Trasformazioni semplici................................................................... 43
2.5.2 Applicazione..................................................................................... 45
2.6 Applicazione ........................................................................................................46
2.7 Esercizi.................................................................................................................49
3 Comportamenti volumetrici dei fluidi puri...................................................................53
3.1 Comportamenti qualitativi....................................................................................54
3.1.1 Applicazione..................................................................................... 61
3.2 Diagrammi di stato e tabelle.................................................................................62
3.3 Relazioni matematiche .........................................................................................63
3.3.1 Equazione di van der Waals e equazioni cubiche............................. 63
3.3.2 Equazione degli stati corrispondenti ................................................ 70
3.3.3 Equazione del viriale........................................................................ 72
3.4 Applicazione ........................................................................................................73
3.5 Esercizi.................................................................................................................86
4 Propriet termodinamiche dei fluidi puri .....................................................................89
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
II
4.1 Funzioni termodinamiche.....................................................................................90
4.1.1 Entalpia ............................................................................................ 92
4.1.2 Energia libera di Helmholtz.............................................................. 93
4.1.3 Energia libera di Gibbs..................................................................... 94
4.1.4 Relazioni di Maxwell ....................................................................... 95
4.2 Principio della minima energia.............................................................................96
4.3 Funzioni residue ...................................................................................................98
4.3.1 EoS del viriale ................................................................................ 103
4.3.1.1 Coefficiente di Joule - Thomson ............................................................104
4.3.2 EoS cubiche.................................................................................... 106
4.3.3 Equazione degli stati corrispondenti .............................................. 109
4.3.4 Applicazione................................................................................... 111
4.4 Diagrammi di stato.............................................................................................119
4.4.1 Applicazione................................................................................... 119
4.5 Fasi condensate ..................................................................................................120
4.5.1 Applicazione................................................................................... 124
4.6 Propriet termodinamiche in condizione di equilibrio di fase............................126
4.7 Fugacit..............................................................................................................131
4.7.1 Condizioni di equilibrio.................................................................. 132
4.7.2 Calcolo della fugacit con equazioni di stato ................................. 133
4.7.3 Fasi condensate .............................................................................. 134
4.7.4 Applicazione................................................................................... 139
4.8 Esercizi...............................................................................................................142
5 Propriet delle miscele ...............................................................................................145
5.1 Propriet delle miscele .......................................................................................146
5.1.1 Propriet parziali molari ................................................................. 148
5.1.1.1 Applicazione...........................................................................................150
5.1.2 Equazione di Gibbs - Duhem......................................................... 151
5.2 Miscele di gas perfetti ........................................................................................151
5.2.1 Propriet di miscelazione ............................................................... 154
5.2.2 Applicazione................................................................................... 155
5.3 Funzioni residue da EoS.....................................................................................156
5.3.1 Equazioni di stato cubiche.............................................................. 158
5.3.2 Equazione del viriale...................................................................... 158
5.3.3 Equazione degli stati corrispondenti .............................................. 160
5.4 Applicazione ......................................................................................................161
5.5 Potenziale chimico e fugacit.............................................................................175
5.5.1 Applicazione................................................................................... 179
5.6 Fasi condensate ..................................................................................................182
5.6.1 Miscele ideali ................................................................................. 182
5.6.1.1 Propriet di miscelazione .......................................................................184
5.6.1.2 Fugacit ..................................................................................................185
5.6.1.3 Applicazione...........................................................................................186
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
III
5.6.2 Miscele non ideali .......................................................................... 188
5.6.2.1 Propriet di miscelazione .......................................................................191
5.6.2.2 Misura dei coefficienti di attivit............................................................191
5.6.2.3 Modelli di coefficienti di attivit............................................................193
5.6.2.4 Applicazione...........................................................................................199
5.7 Esercizi...............................................................................................................201
6 Equilibrio tra fasi........................................................................................................203
6.1 Regola delle fasi di Gibbs ..................................................................................204
6.2 Condizioni di equilibrio .....................................................................................205
6.3 VLE....................................................................................................................206
6.3.1 Calcolo del punto di bolla e di rugiada........................................... 215
6.3.2 Bassa e media pressione: metodi indiretti (approccio /) ............. 215
6.3.2.1 Punto di bolla .........................................................................................218
6.3.2.2 Punto di rugiada......................................................................................220
6.3.2.3 Applicazione...........................................................................................221
6.3.2.4 Composti supercritici .............................................................................227
6.3.3 Alta pressione: metodi diretti (approccio /)................................ 231
6.3.4 Applicazione................................................................................... 233
6.4 LLE ....................................................................................................................236
6.4.1 Calcolo dello stato di equilibrio a media e bassa pressione............ 244
6.4.2 Applicazione................................................................................... 245
6.4.3 VLLE con composti in fase liquida completamente immiscibili a bassa e
media pressione.............................................................................................. 247
6.4.3.1 Applicazione...........................................................................................250
6.5 SLE.....................................................................................................................253
6.5.1.1 Applicazione...........................................................................................256
6.6 Leggi limite per sistemi diluiti ...........................................................................260
6.6.1 Abbassamento del punto di congelamento ..................................... 260
6.6.2 Abbassamento della tensione di vapore.......................................... 262
6.6.3 Innalzamento della temperatura di ebollizione............................... 263
6.6.4 Ripartizione di un soluto in due solventi ........................................ 264
6.6.5 Solubilit dei gas nei liquidi ........................................................... 265
6.7 Esercizi...............................................................................................................265
7 Sistemi reagenti..........................................................................................................269
7.1 Bilancio di materia in sistemi multicomponente ................................................270
7.1.1 Sistemi non reagenti ....................................................................... 272
7.2 Sistemi chiusi .....................................................................................................273
7.2.1 Il vincolo stechiometrico................................................................ 273
7.2.2 Il grado di avanzamento per sistemi semplici................................. 276
7.2.3 Condizione di equilibrio per sistemi semplici ................................ 279
7.2.4 Stati di riferimento standard........................................................... 283
7.2.4.1 Composti in fase gas nello stato del sistema ..........................................283
7.2.4.2 Composti in fase liquida nello stato del sistema.....................................284
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
IV
7.2.4.3 Composti in fase solida nello stato del sistema ......................................285
7.2.5 Influenza della temperatura sulla costante di equilibrio................. 285
7.2.6 Applicazione................................................................................... 288
7.2.7 Sistemi complessi ........................................................................... 293
7.2.8 Applicazione................................................................................... 295
7.3 Sistemi aperti in condizioni stazionarie..............................................................297
7.3.1 Applicazione................................................................................... 299
7.4 Bilancio di energia in sistemi multicomponente ................................................301
7.4.1 Sistemi chiusi ................................................................................. 301
7.4.2 Sistemi aperti in condizioni stazionarie.......................................... 304
7.4.3 Applicazione................................................................................... 305
7.5 Esercizi...............................................................................................................313
8 Appendice A Richiami di matematica........................................................................317
8.1 Relazioni fondamentali ......................................................................................317
8.1.1 Logaritmi ed esponenziali .............................................................. 317
8.1.2 Equazioni di secondo grado............................................................ 317
8.1.3 Vettori e matrici ............................................................................. 317
8.1.4 Derivate, differenziali ed integrali.................................................. 318
8.1.5 Approssimazioni ed interpolazioni................................................. 319
8.1.5.1 Espansione in serie di Taylor .................................................................320
8.1.5.2 Interpolazione lineare.............................................................................320
8.1.5.3 Problematiche connesse col calcolo numerico .......................................321
8.2 Differenziali esatti e funzioni di stato ................................................................323
8.3 Sistemi con due variabili indipendenti ...............................................................324
9 Appendice B Tabelle termodinamiche dellacqua .....................................................327
10 Appendice C Tabelle di Lee - Kesler .....................................................................335
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
V
Prefazione
Questo testo stato scritto per gli studenti del corso di Laurea in Ingegneria Chimica del
Politecnico di Milano e la sua organizzazione risente quindi della struttura locale del Corso
di Studio. Nel Nuovo Ordinamento degli Studi del Politecnico di Milano i contenuti di
Termodinamica dellIngegneria Chimica sono stati suddivisi in due corsi da cinque crediti
ciascuno: il primo corso ha trovato collocazione al secondo anno del corso di Laurea ed il
secondo corso al primo anno del corso di Laurea Specialistica. Come corsi propedeutici gli
studenti del corso di Laurea seguono al primo anno un corso di Fisica Generale ed uno di
Fisica Tecnica, oltre ai corsi di base di Analisi Matematica. Pur assumendo che lo studente
abbia seguito con profitto i corsi propedeutici di Fisica Generale e Fisica Tecnica, il primo
capitolo riporta alcuni brevi richiami sui sistemi termodinamici, le trasformazioni
spontanee e le condizioni di equilibrio. Questo rende il testo fruibile anche da studenti di
corsi di Laurea che non prevedano la frequenza di tali corsi propedeutici. Inoltre i principali
strumenti matematici necessari alla comprensione del materiale esposto nel testo sono
riassunti in Appendice A.
Il presente testo si rivolge quindi principalmente agli studenti del secondo anno del corso di
Laurea ed stato scritto cercando di soddisfare due esigenze contrastanti: da un lato esporre
i fondamenti della Termodinamica dellIngegneria Chimica con un taglio semplice ed
applicativo (compatibile quindi con gli obiettivi formativi di un corso di Laurea triennale),
dallaltro fornire agli studenti pi motivati materiale sufficiente per approfondire lo studio.
Si sono quindi sacrificati molti interessanti approfondimenti che vengono trattati solo
brevemente nel testo, ma si cercato di segnalare riferimenti a testi o ad articoli pubblicati
su riviste scientifiche dove lo studente che lo desidera pu facilmente trovare delle chiare
esposizioni degli aspetti non approfonditi nel testo.
La Termodinamica dellIngegneria Chimica si differenzia dalla Termodinamica Classica in
quanto il suo ambito di indagine principale sono i sistemi multicomponente, cio le
miscele. Ci nonostante, la struttura del testo premette la trattazione della termodinamica
dei composti puri a quella delle miscele, in quanto lo studio di queste ultime poggia su
metodologie sviluppate per i fluidi puri. Lobiettivo finale quello di fornire allo studente
dei metodi di calcolo delle propriet volumetriche e termodinamiche e dello stato di
equilibrio di sistemi contenenti un numero qualsiasi di specie chimiche.
La parte relativa ai fluidi puri trattata quindi nei primi capitoli: dopo un breve richiamo
dei fondamenti della Termodinamica Classica riportato nel capitolo 1, il secondo capitolo
formalizza la struttura dei bilanci di materia, energia ed entropia da cui si possono ricavare
le note leggi della termodinamica come casi particolari. Il terzo capitolo affronta il
problema della previsione, attraverso luso di equazioni di stato di diversa complessit,
delle propriet volumetriche. Le equazioni di stato introdotte nel terzo capitolo vengono poi
utilizzate nel quarto capitolo per il calcolo delle altre propriet termodinamiche (energia
interna, entalpia, entropia, energia libera di Gibbs, fugacit, ) dei fluidi puri.
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
VI
I rimanenti capitoli affrontano il problema delle miscele. Il capitolo 5 estende i concetti
affrontati nei capitoli 3 e 4 al caso del calcolo delle propriet volumetriche e
termodinamiche delle miscele, mentre i capitoli 6 e 7 affrontano il problema della
definizione dello stato di equilibrio di un sistema contenente un numero arbitrario di fasi e
di specie chimiche. In particolare, il capitolo 6 affronta il caso dellequilibrio tra fasi in
assenza di trasformazioni chimiche (equilibrio liquido vapore, liquido liquido, liquido
liquido vapore e solido liquido), mentre il capitolo 7 rimuove la limitazione
dellassenza di trasformazioni chimiche per arrivare alla formalizzazione dei bilanci di
materia ed energia nel caso pi generale di sistemi multicomponente reagenti in condizioni
di equilibrio termodinamico.
Per rendere il testo semplice ed applicativo si cercato di presentare la materia secondo un
approccio sperimentale (piuttosto che assiomatico) e si corredato il testo di numerose
applicazioni numeriche completamente risolte. Inoltre, al termine di ciascun capitolo sono
proposti degli esercizi di cui si fornisce solo il risultato numerico al fine di consentire allo
studente una verifica autonoma della comprensione del materiale esposto nel capitolo.
Il testo riporta anche i listati di programmi in un linguaggio evoluto (Matlab
) per la
risoluzione delle applicazioni proposte: lutilizzo di linguaggi evoluti su personal computer
riveste per lo studente attuale lo stesso ruolo che ha rivestito lutilizzo della calcolatrice
tascabile al posto del regolo calcolatore per gli studenti degli anni 70 del secolo scorso.
Linserimento di listati commentati vuole essere uno stimolo per lo studente ad imparare ed
utilizzare nella pratica quotidiana questi linguaggi di programmazione evoluti al posto della
calcolatrice tascabile. Inoltre, i programmi forniti possono costituire la base per la
risoluzione, attraverso semplici modifiche, di un gran numero di problemi tipici che
lingegnere chimico si trova ad affrontare nella pratica professionale.
Nonostante lo studio della materia esposta in questo testo possa apparire a prima vista
molto impegnativo, in realt esso si basa su un gran numero di applicazioni diverse di pochi
concetti (ed equazioni) fondamentali. Si suggerisce quindi allo studente di cercare di
cogliere le molte analogie tra gli argomenti esposti, evitando uno sterile esercizio di pura
memorizzazione delle molte equazioni riportate. Per quanto probabilmente superfluo, si
ricorda inoltre linutilit di affrontare le applicazioni e gli esercizi riportati senza aver prima
studiato e compreso la teoria che essi vogliono illustrare. Non sar inoltre mai sottolineata a
sufficienza limportanza di risolvere autonomamente gli esercizi proposti fino al risultato
numerico finale, come unico strumento di reale valutazione del livello di preparazione
raggiunto.
Numerosi refusi sono stati eliminati grazie al lavoro degli studenti che hanno dovuto
preparare lesame sulle bozze di questo testo. Ci nonostante assai probabile che altri
refusi siano presenti in questa prima edizione: si ringraziano anticipatamente tutti coloro
che vorranno segnalarli.
Renato Rota
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
VII
Nomenclatura
a accelerazione [m s
-2
]
a coefficiente energetico delle EoS cubiche [Pa m
6
kmol
-2
]
a attivit
A superficie [m
2
]
A energia libera di Helmholtz [J]
A matrice [a
ki
] specie elementi
A, B coefficienti delle EoS cubiche
A, B, C costanti della equazione di Antoine
A
12
, A
21
costanti dellequazione di van Laar
a
ki
numero di atomi di tipo k nella molecola di tipo i
b covolume [m
3
kmol
-1
]
B secondo coefficiente del viriale [m
3
kmol
-1
]
B costante nel modello di Margules
B secondo coefficiente del viriale [Pa
-1
]
b
k
numero di moli di atomi di tipo k presenti inizialmente [mol]
C capacit termica o calore specifico [J K
-1
]
C terzo coefficiente del viriale [m
6
kmol
-2
]
C terzo coefficiente del viriale [Pa
-2
]
c
p
calore specifico molare a pressione costante [J mol
-1
K
-1
]
P
c
calore specifico massico a pressione costante [J kg
-1
K
-1
]
C
p
calore specifico a pressione costante [J K
-1
]
c
V
calore specifico molare a volume costante [J mol
-1
K
-1
]
V
c
calore specifico massico a volume costante [J kg
-1
K
-1
]
C
V
calore specifico a volume costante [J K
-1
]
D discriminante della EoS cubica
E energia totale del sistema [J]
E vettore degli elementi
E
&
flusso di energia associato alle portate materiali [W]
k
E energia cinetica [J]
k
E
&
flusso di energia cinetica [W]
p
E energia potenziale [J]
p
E
&
flusso di energia potenziale [W]
f fugacit [Pa]
i
f
fugacit del composti i-esimo in miscela [Pa]
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
VIII
F forza [N]
F numero di fasi
g accelerazione di gravit [m s
-2
]
g energia libera di Gibbs molare [J mol
-1
]
G energia libera di Gibbs [J]
g
c
fattore dimensionale nel Sistema Anglosassone [lb
m
ft lb
f
-1
s
-2
]
G
i
produzione molare della specie i [mol]
i
G
&
velocit di produzione molare della specie i [mol s
-1
]
i
G energia libera di Gibbs parziale molare [J mol
-1
]
h entalpia molare [J mol
-1
]
h
entalpia massica [J kg
-1
]
H entalpia [J]
H costante di Henry [Pa]
i
H entalpia parziale molare [J mol
-1
]
i
H
&
flusso di entalpia [W]
k parametro delle EoS cubiche
K rapporto di vaporizzazione
K costante di equilibrio
i
K
coefficiente di ripartizione di Nernst della specie i-esima tra le due
fasi e
L spostamento [m]
m massa [kg]
m& portata massica [kg s
-1
]
M peso molecolare
n numero di moli [mol]
n&
portata molare [mol s-1]
N numero di specie
NA numero di relazioni che si originano dal vincolo stechiometrico
NR numero di relazioni stechiometriche
P pressione [Pa]
P tensione di vapore [Pa]
P
i
pressione parziale [Pa]
P
C
pressione critica [Pa]
o
R
P
tensione di vapore ridotta
q calore molare [J mol
-1
]
Q calore [J]
Q
&
potenza termica [W]
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
IX
Q
calore fornito per kg di materia fluente dal sistema [J kg
-1
]
R costante dei gas perfetti [J mol
-1
K
-1
]
S
R
&
generazione di entropia [W K
-1
]
S
R
generazione di entropia per kg di materia fluente nel sistema [J kg
-1
K
-1
]
s entropia molare [J K
-1
mol
-1
]
s entropia massica [J K
-1
kg
-1
]
S entropia [J K
-1
]
S parametro delle EoS cubiche
S vettore delle specie
i
S entropia parziale molare [J mol
-1
K
-1
]
S
&
flusso di entropia [W K
-1
]
t tempo [s]
T
temperatura [K]
T
C
temperatura critica [K]
T
R
temperatura ridotta [K]
u energia interna molare [J mol
-1
]
u energia interna massica [J kg
-1
]
U energia interna [J]
i
U energia interna parziale molare [J mol
-1
]
U
&
flusso di energia interna [W]
u, w coefficienti delle relazioni generali per le EoS cubiche
v velocit [m s
-1
]
v volume molare [m
3
mol
-1
]
v volume massico [m
3
kg
-1
]
V volume [m
3
]
V varianza
v
C
volume molare critico [m
3
mol
-1
]
i
V volume parziale molare [m
3
mol
-1
]
W lavoro [J]
W
&
potenza meccanica [W]
x titolo in vapore [kg
V
kg
-1
]
x frazione molare [mol
i
mol
tot
-1
]
y frazione molare [mol
i
mol
tot
-1
]
z
quota [m]
Z coefficiente di compressibilit
Z
C
coefficiente di compressibilit critico
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
X
Lettere greche
, , coefficienti delle EoS cubiche
rapporto tra
*
P
c e
*
V
c
coefficiente di attivit
0
R
G energia libera di Gibbs di reazione [J mol
-1
]
0
R
H entalpia di reazione [J mol
-1
]
variazione finita
grado di avanzamento [mol]
&
grado di avanzamento [mol s
-1
]
potenziale chimico [J mol
-1
]
coefficiente di Joule Thomson [K Pa
-1
]
coefficiente stechiometrico
densit molare [mol m
-3
]
densit massica [kg m
-3
]
R
densit ridotta
coefficiente di fugacit
i
coefficiente di fugacit del composti i-esimo in miscela
fattore acentrico di Pitzer
Apici
miscela ideale
* gas perfetto
0 stato iniziale
0 in ingresso
0 stato standard
A, B, sottosistema A, B,
E di eccesso
f di formazione
L liquido
mix di miscelazione
R residua
S solido
V vapore
x convenzione asimmetrica
a diluizione infinita
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
XI
, , fase , ,
Pedici
1, 2, stati di equilibrio di un sistema
ad adiabatico
b di bolla
C critica
cond di condensazione
d di rugiada
E di eutettico
ev di evaporazione
f, fus di fusione
i, j, della specie i, j,
IN della corrente in ingresso
k della reazione k
OUT della corrente in uscita
r di riferimento
R ridotta
rel relativa
Simbologia generale
f(x,y) generica grandezza f funzione delle variabili x,y
m grandezza generica molare
m vettore della grandezza m
m grandezza generica massica
M grandezza generica
i
M grandezza generica parziale molare rispetto alla specie i-esima
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
1
1 Definizioni e concetti propedeutici
In questo capitolo verranno brevemente richiamati alcuni concetti di base che si
assumono gi noti, nella forma qui presentata o in altre forme equivalenti, allo studente.
Al termine dello studio di questo capitolo lo studente dovrebbe conoscere:
il significato e la validit di una legge termodinamica;
il significato concettuale di equilibrio termodinamico;
la definizione di sistema termodinamico, di ambiente e di superficie di controllo;
i sistemi aperti, chiusi e isolati;
la differenza tra grandezze intensive, estensive e specifiche;
il Sistema Internazionale e quello Anglosassone di unit di misura;
il concetto di stato di un sistema e di funzione di stato;
il concetto di lavoro, lavoro meccanico e calore;
il significato di trasformazione reversibile e spontanea;
il concetto di energia cinetica, potenziale e interna;
lanalisi dimensionale;
il concetto di entropia;
le condizioni di equilibrio al trasferimento di calore, energia meccanica e materia.
1.1 Leggi della termodinamica
La termodinamica una scienza nata nel diciannovesimo secolo dalla necessit di
descrivere in modo quantitativo i limiti di funzionamento dei motori a vapore. Il nome
stesso deriva dallidea di trasformare calore (termo) in potenza meccanica (dinamica).
Essa quindi nata per occuparsi dellenergia e delle sue trasformazioni. I principi
osservati sperimentalmente sono stati formalizzati in relazioni matematiche che
costituiscono le cosiddette leggi della termodinamica. Queste leggi non hanno una
dimostrazione matematica in senso stretto, ma basano la loro validit sullassenza di
evidenze sperimentali che le contraddicono.
Le leggi della termodinamica portano, attraverso delle relazioni matematiche esatte, a
un insieme di relazioni che hanno trovato vasta applicazione praticamente in tutte le
scienze applicate, compresa quindi lingegneria chimica.
Partendo da queste leggi nate dallo studio dei motori a vapore infatti possibile
caratterizzare in modo quantitativo fenomeni completamente diversi, come per esempio
la quantit di calore necessaria per fare avvenire un certo processo chimico o le
condizioni di equilibrio a cui giunge una reazione chimica.
Con condizioni di equilibrio si intendono quelle condizioni che una certa quantit di
materia, soggetta a determinati vincoli (per esempio quello di non variare il valore della
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
2
temperatura e della pressione), raggiunge dopo un certo tempo (eventualmente infinito)
e da cui non ha pi alcuna tendenza a spostarsi.
Queste condizioni (di equilibrio) rappresentano il campo di applicazione delle leggi
della termodinamica. In altri termini, la termodinamica ci dice dove andiamo a finire,
non come o con che velocit. La termodinamica ci dice per esempio se certi composti
chimici reagiscono spontaneamente per formare altri composti, e la quantit di questi
composti che si formano nelle condizioni di equilibrio. Non ci dice se questa
trasformazione avviene in un secondo o in diecimila anni.
1.2 Sistemi termodinamici
Tutte le applicazioni delle leggi della termodinamica a un problema reale iniziano con la
definizione di una certa quantit di materia su cui viene focalizzata lattenzione. Questa
quantit di materia viene chiamata sistema, mentre tutto ci che non appartiene al
sistema viene chiamato ambiente. Linsieme unione del sistema e dellambiente
quindi luniverso. La definizione univoca di un sistema richiede la definizione di una
superficie di controllo, reale o immaginaria, che ne delimita i confini. La materia che si
trova allinterno della superficie di controllo rappresenta il sistema, quella esterna
lambiente.
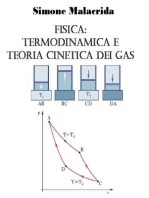
Figura 1: esempi di differenti scelte della superficie di controllo.
LIQUIDO
VAPORE
S1
S2
S3
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
3
Dato un problema, la definizione della superficie di controllo arbitraria, ma una scelta
oculata solitamente facilita notevolmente la risoluzione del problema. Alcuni esempi di
scelta della superficie di controllo per un semplice separatore di fase
1
vapore e liquida
con uno scambiatore di calore tra la corrente gassosa uscente e quella entrante
mostrato in Figura 1. La superficie di controllo S1 racchiude entrambe le
apparecchiature, la S2 solo il separatore di fase con la valvola sulla linea di
alimentazione, mentre la S3 racchiude solo lo scambiatore di calore.
Una superficie di controllo pu essere attraversata da flussi
2
di materia e/o di energia.
Per esempio, la superficie di controllo S1 in Figura 1 attraversata da un flusso
materiale in ingresso e da due flussi materiali (luno liquido e laltro vapore) in uscita.
Questi flussi definiscono ci che entra (se diretti verso linterno) e ci che esce (se
diretti verso lesterno) dal sistema.
Una superficie di controllo (o una sua parte) si dice impermeabile se non consente il
passaggio di flussi di materia e permeabile viceversa. Analogamente, viene chiamata
adiabatica se non consente il passaggio di flussi di calore e diatermana se lo consente.
Infine, una superficie di controllo viene chiamata mobile se consente il passaggio di
flussi di lavoro e rigida se lo impedisce.
Un sistema delimitato da una superficie di controllo impermeabile, rigida e adiabatica
non pu scambiare n materia n energia con lambiente e si dice isolato. Se la
superficie di controllo impermeabile, ma diatermana e/o mobile il sistema pu
scambiare energia con lambiente ma non materia e viene chiamato chiuso. Un sistema
aperto pu scambiare con lambiente sia materia sia energia ed quindi delimitato da
una superficie di controllo permeabile, eventualmente anche diatermana e/o mobile.
Non pu esistere un sistema che scambia materia con lambiente senza scambiare
energia in quanto un flusso di materia porta con s inevitabilmente un flusso di energia.
1.3 Propriet termodinamiche e unit di misura
Lo stato di un sistema viene definito dal valore che assumono le sue propriet
termodinamiche. Come verr ampiamente discusso nel seguito, alcune di queste
propriet sono misurabili (come temperatura, pressione, quantit di materia o volume)
mentre altre no. Queste ultime possono per essere calcolate a partire dai valori delle
propriet misurabili attraverso delle relazioni matematiche.
1
Con separatore di fase si intende unapparecchiatura in grado di separare due fasi, per esempio
una fase liquida da una fase vapore.
2
Con flusso si intende la quantit (di materia o energia) che attraversa una superficie nellunit di
tempo.
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
4
In particolare, come detto in precedenza, la termodinamica si occupa degli stati di
equilibrio. Gli stati di equilibrio sono ovviamente enormemente pi semplici da
descrivere di quelli in cui sono in atto delle trasformazioni; tale semplicit si riflette
nella possibilit di descrivere gli stati di equilibrio con un numero limitato di propriet
termodinamiche.
Una propriet termodinamica si dice estensiva se il suo valore dipende dalla
estensione del sistema, cio dalla quantit di materia in esso contenuta. Viceversa, se
il suo valore non dipende dalla quantit di materia contenuta nel sistema la propriet
viene chiamata intensiva. Per esempio, il volume una propriet (o grandezza: i due
termini verranno usati come sinonimi) estensiva mentre la temperatura una grandezza
intensiva. Per le grandezze estensive vale la propriet additiva, mentre per le grandezze
intensive no. Per esempio, il sistema che risulta dallunione di due sistemi con volume
V
A
il primo e V
B
il secondo ha un volume V=V
A
+V
B
. Viceversa, il sistema che risulta
dallunione di due sistemi con temperatura pari a T
A
il primo e a T
B
il secondo non ha
una temperatura T=T
A
+T
B
.
Un sistema termodinamico pu evolvere attraverso delle trasformazioni, cio dei
cambiamenti successivi del suo stato. Le trasformazioni si dicono isoterme se la
temperatura rimane costante, isobare se rimane costante la pressione, isocore se rimane
costante il volume, adiabatiche se non vi flusso di calore attraverso la superficie di
controllo.
Se il valore che assume una grandezza termodinamica dipende solo dallo stato del
sistema e non dalle trasformazioni che il sistema stesso ha subito in precedenza si dice
che tale grandezza una funzione di stato. Ne consegue che se un sistema subisce una
trasformazione da uno stato iniziale a uno finale, la conseguente variazione del valore di
una sua funzione di stato dipende solo dallo stato iniziale e da quello finale e non dalla
trasformazione subita. Matematicamente, questo implica che il differenziale di queste
funzioni deve essere un differenziale esatto.
Se si considerano solo le variabili intensive si parla di stato intensivo del sistema.
Sperimentalmente, si verifica che lo stato intensivo di un sistema monofase
3
e
monocomponente completamente definito quando sono fissate due variabili intensive
4
.
In altri termini, se partendo da un certo stato intensivo si compiono delle trasformazioni
3
Si ricorda che una fase una porzione di materia con caratteristiche omogenee. Un sistema
eterogeneo formato da pi fasi: attraversando la superficie di separazione tra due fasi si riscontra
una brusca variazione di almeno una propriet intensiva del sistema (per esempio, la densit). Una
miscela di liquido e vapore un sistema eterogeneo costituito da due fasi: luna liquida, laltra
vapore. Quindi, i termini sistema monofase e sistema omogeneo verranno utilizzati come
sinonimi.
4
Questo vero per i problemi in cui possono essere trascurati gli effetti di campo (magnetico,
elettrico, ), di superficie e gli sforzi viscosi.
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
5
tali da riportare due variabili intensive al loro valore iniziale, anche tutte le altre
variabili intensive ritornano al loro valore iniziale.
Il valore assunto da una qualsiasi propriet termodinamica definito da ununit di
misura, che indica quale lo standard utilizzato per misurare quella propriet, e da un
numero, che definisce quante unit standard servono per raggiungere il valore della
propriet considerata. Per esempio, la pressione di un sistema pari a 100 (numero) bar
(unit di misura). Le unit di misura sono definite allinterno di diversi sistemi e
verranno indicate nel seguito tra parentesi quadre (per esempio, P = 3 [bar]).
Ad ogni propriet estensiva corrisponde una propriet specifica, definita come il valore
della propriet associata allunit di massa (propriet massiche) o di materia (propriet
molari). Per esempio, al volume, misurato in [m
3
], corrisponde il volume massico, cio
il volume occupato dallunit di massa e misurato in [m
3
/kg], e il volume molare, cio il
volume occupato dallunit di materia e misurato in [m
3
/mol]. Questi sono linverso
delle pi note densit, rispettivamente massica e molare.
Le grandezze specifiche sono per loro natura intensive e si indicheranno nel seguito con
la lettera minuscola, senza altra specificazione le molari e soprassegnate le massiche.
Cos si indicher con V [m
3
] il volume, con v [m
3
/mol] il volume molare e con v [m
3
/kg]
il volume massico.
1.3.1 Sistema Internazionale
Il Sistema Internazionale di unit di misura (abbreviato con SI, dal francese Systme
International) definisce come primitive (almeno per quanto di interesse per la
termodinamica dellingegneria chimica) cinque grandezze misurabili ed assegna ad esse
le relative unit di misura:
tempo, con unit di misura secondo [s];
lunghezza, con unit di misura metro [m];
temperatura, con unit di misura kelvin [K];
massa, con unit di misura chilogrammo [kg];
quantit di materia, con unit di misura mole [mol].
Il valore di queste grandezze primitive assegnato per definizione e non pu essere
quindi dedotto da grandezze pi semplici. Sulla base di leggi fisiche, dalle unit di
misura delle grandezze primitive si deducono quelle delle grandezze derivate, alcune
delle quali sono discusse nel seguito.
La forza il prodotto della massa per laccelerazione (seconda legge del moto di
Newton
5
):
5
Isaac Newton, 1642 - 1727, matematico, scienziato e filosofo inglese.
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
6
] [ ] [
2
= ms a kg m F (1)
da cui si deduce che lunit di misura della forza [kg m s
-2
], anche chiamata newton,
[N].
Larea il prodotto di due lunghezze, e quindi si misura in [m
2
], mentre il volume il
prodotto di tre lunghezze, e quindi si misura in [m
3
].
La pressione la forza esercitata da un fluido normalmente a una superficie per unit di
area:
] [
] [
2
m A
N F
P =
(2)
e quindi lunit di misura della pressione [N m
-2
], anche chiamata pascal, [Pa]. 1 [bar]
equivale a 10
5
[Pa].
Il lavoro lazione della componente di una forza che agisce nella direzione di uno
spostamento:
] [ ] [ m dL N F dW = (3)
dove si considerato uno spostamento infinitesimo (dL) che quindi origina un lavoro
anchesso infinitesimo (dW). Lunit di misura del lavoro quindi [N m], anche
chiamata joule, [J]. Per convenzione, si considera positivo il lavoro quando forza e
spostamento hanno lo stesso verso.
Figura 2: lavoro di espansione di un gas (si noti che la superficie di controllo contiene
tutto il gas presente nel cilindro e varia nel tempo).
SUPERFICIE DI
CONTROLLO
SPOSTAMENTO
PRESSIONE
INTERNA
PRESSIONE
ESTERNA
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
7
Un tipo di lavoro incontrato spesso in termodinamica quello di tipo meccanico, legato
alla espansione o alla compressione di un fluido come illustrato in Figura 2. Nelle
condizioni iniziali il sistema (definito dalla superficie di controllo che include tutto il
gas presente nel cilindro) si trova alla pressione P
1
e occupa il volume V
1
. Poich il
sistema si trova in condizioni di equilibrio, la pressione interna uguale a quella
esterna, altrimenti la risultante delle forze agenti sul pistone non sarebbe nulla e il
pistone tenderebbe a muoversi.
Se si riduce la pressione esterna di una quantit infinitesima, il pistone si muover verso
destra in modo infinitamente lento e il gas si espander sempre in modo infinitamente
lento. La pressione interna e quella esterna saranno sempre praticamente uguali, a meno
di una quantit infinitesima. Pur espandendosi, il sistema in condizioni di quasi
equilibrio, nel senso che lo scostamento dalle condizioni di equilibrio infinitesimo. La
trasformazione viene detta reversibile
6
e termina dopo uno spostamento infinitesimo del
pistone, dL. Espandendosi, il sistema compie un lavoro contro la forza esercitata sul
pistone dalla pressione esterna che, in ogni istante, uguale a quella interna essendo la
trasformazione reversibile. Tale forza quindi uguale allarea del pistone per la
pressione del sistema, e il lavoro infinitesimo uscente dal sistema (cio che il sistema
compie sullambiente durante lespansione) sar negativo (in quanto forza e
spostamento hanno verso discorde) e pari a:
PdV PAdL FdL dW = = = (4)
Il segno meno tiene conto della convenzione di segno per il lavoro. Se il sistema si
espande, forza e spostamento hanno verso discorde e il lavoro deve essere negativo.
Poich per dV positivo, anche il prodotto PdV risulta positivo e il segno meno serve a
rendere negativo il lavoro. Se il sistema viene compresso, forza e spostamento hanno lo
stesso verso e il lavoro deve essere positivo. In questo caso per dV negativo, il
prodotto PdV risulta anchesso negativo e il segno meno rende positivo il lavoro. Se
lespansione procede per stadi di quasi equilibrio fino al valore finale di volume V
2
, il
lavoro fatto dal sistema pari a:
= =
2
1
2
1
V
V
PdV dW W (5)
Questa relazione, importante ricordarlo, vale solo per trasformazioni reversibili e pu
essere visualizzata su un piano cartesiano P - V come mostrato in Figura 3. Lintegrale
presente nella relazione (5) pari allarea sottesa alla curva che rappresenta la
trasformazione sul diagramma di Figura 3, dove sono riportate a titolo di esempio due
6
In quanto una variazione infinitesima di segno opposto della pressione porta il sistema a
ripercorrere esattamente la stessa trasformazione in senso inverso, cosa generalmente non vera per
trasformazioni che evolvono con una velocit finita.
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
8
diverse trasformazioni. immediato comprendere che, pur essendo uguali gli stati
iniziali e finali, il lavoro relativo alle due trasformazioni diverso. Ne consegue che il
lavoro non una funzione di stato e il suo differenziale non esatto.
Il lavoro, come anche il calore di cui parleremo di seguito, non una forma di energia
che pu essere accumulata in un sistema, ma rappresenta una forma di energia che viene
scambiata tra il sistema e lambiente. In altri termini, si tratta di energia in transito
attraverso la superficie di controllo.
Lequivalenza tra calore e lavoro stata messa in luce dal celebre esperimento di Joule
7
effettuato con lapparecchiatura mostrata in Figura 4. Fornendo al fluido presente in un
recipiente coibentato del lavoro (sotto forma di moto delle pale immerse nel fluido
stesso, moto generato dallabbassamento della quota di una certa massa) la temperatura
del fluido aumenta. Se poi si rimuove il coibente dalle pareti del recipiente e gli si
consente di scambiare calore con un corpo pi freddo, la temperatura del sistema si
riporta al valore iniziale mentre il corpo pi freddo si riscalda.
Figura 3: lavoro di espansione.
7
James Prescott Joule, 1818 - 1889, fisico inglese.
P
P
1
P
2
V
1
V
2
V
STATO
INIZIALE
STATO
FINALE
TRASFORMAZIONE A
TRASFORMAZIONE B
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
9
Da questa esperienza sono evidente due fatti: da un lato, lavoro e calore sono due forme
di energia in transito attraverso la superficie di controllo di un sistema che possono
essere convertite luna nellaltra; dallaltro, possibile immagazzinare dellenergia in
un sistema fermo.
Da queste evidenze conseguono immediatamente altre considerazioni.
La prima e pi ovvia che anche il calore (essendo una forma di energia in transito
attraverso la superficie di controllo come il lavoro) si misura in joule.
La seconda invece pi sottile: anche un corpo in quiete pu variare la sua energia.
Questo non affatto ovvio considerando le forme classiche dellenergia meccanica
(quella cinetica e quella potenziale, che erano le uniche considerate nel principio di
conservazione dellenergia meccanica formulato alla fine del XVII secolo da Leibnitz
8
),
la cui variazione sempre legata allo spostamento del sistema.
Figura 4: apparecchiatura utilizzata nellesperienza di Joule.
8
Gottfried Wilhelm von Leibnitz, 1646 - 1716, filosofo e matematico tedesco.
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
10
Lenergia cinetica infatti legata alla variazione di velocit di un sistema. In generale il
lavoro viene espresso dalla relazione (3); considerando che per un corpo in moto che
varia la sua velocit la forza pari alla massa per laccelerazione, e che laccelerazione
pari alla derivata della velocit nel tempo, si ottiene:
mvdv dv
dt
dL
m dL
dt
dv
m madL FdL dW = = = = = (6)
e quindi il lavoro compiuto da un corpo che cambia la sua velocit da un valore v
1
a un
valore v
2
pari a:
k
E
mv mv
mvdv W = = =
2 2
2
1
2
2
2
1
(7)
dove con si indicata una variazione finita e con E
k
, secondo la notazione introdotta
da Kelvin
9
, lenergia cinetica.
Analogamente, per variare la quota di un sistema da z
1
a z
2
necessario compiere un
lavoro contro la forza peso (se si aumenta la quota) pari a:
p
E z z mg mgL FL W = = = = ) (
1 2
(8)
dove con E
p
si indicata, secondo la notazione introdotta da Rankine
10
, lenergia
potenziale.
Nellesperimento di Joule il sistema rimane fermo, e quindi nessuna di queste due forme
di energia varia e pu giustificare laccumulo di energia allinterno del sistema quando
si fornisce lavoro, energia che poi viene restituita sotto forma di calore quando si
rimuove la coibentazione.
Risulta quindi necessario ipotizzare lesistenza di una differente forma di energia,
chiamata energia interna, che non dipende dal moto del sistema nel suo insieme, ma dal
moto delle particelle presenti nel sistema
11
. Lenergia interna quindi lenergia che un
sistema possiede intrinsecamente per il fatto che costituito da molecole e atomi in
movimento ed una funzione di stato.
9
Lord William Thomson Kelvin, 1824 - 1907, matematico e fisico irlandese.
10
William John Macquorn Rankine, 1820 1872, scienziato scozzese.
11
Pi precisamente, dai diversi moti delle molecole (energia traslazionale, dovuta al movimento
della molecola nel suo insieme, e rotazionale, dovuta al moto rotazionale della molecola attorno
al suo centro di massa), degli atomi che le costituiscono (energia vibrazionale, dovuta agli
spostamenti degli atomi attorno alle loro posizioni di equilibrio), degli elettroni e dei nuclei che
costituiscono gli atomi. Inoltre, vi lenergia associata alle interazioni tra le diverse molecole.
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
11
Infine, il flusso di energia viene chiamato potenza e si misura in [J s
-1
], detto anche watt,
[W].
1.3.2 Analisi dimensionale
Qualsiasi relazione matematica utilizzata nellambito dellingegneria deve essere
dimensionalmente consistente, nel senso che le unit di misura di tutti gli addendi
devono essere le stesse.
Per esempio, nella relazione
|
|
.
|
\
|
=
2
] [
2
] [
] [
2 1 2
1
2 1 2
2
ms v ms v
kg m W (9)
entrambe le velocit devono essere espresse in [m s
-1
] e il lavoro risulta espresso in [kg]
[m s
-1
]
2
= [kg m
2
s
-2
] = [J].
Analogamente, nella relazione
] [ ] [ ] [ ] [ ] [
3
K T R mol n m V Pa P K = (10)
lunit di misura di R [Pa m
3
mol
-1
K
-1
] = [N m
-2
m
3
mol
-1
K
-1
] = [J mol
-1
K
-1
].
1.3.3 Sistema Anglosassone
Per quanto sia ormai in via di estinzione, questo sistema di unit di misura sopravvive
ancora nella manualistica tecnica di matrice anglosassone. Risulta quindi necessario
conoscerne almeno i rudimenti.
Il Sistema Anglosassone definisce come primitive (almeno per quanto di interesse per la
termodinamica dellingegneria chimica) sei grandezze misurabili ed assegna ad esse le
relative unit di misura:
tempo, con unit di misura secondo [s];
lunghezza, con unit di misura piede [ft];
temperatura, con unit di misura gradi rankine [R];
massa, con unit di misura libbra massa [lb
m
];
quantit di materia, con unit di misura libbra mole [lb
mol
];
forza, con unit di misura libbra forza [lb
f
].
Il fatto di considerare come primitiva anche la forza pone per il problema di rendere
consistente dimensionalmente la seconda legge del moto di Newton, che deve essere
scritta come:
] [ ] [ ] [ ] [
2
= fts a lb m lb F g
m f c
K (11)
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
12
dove g
c
rappresenta il fattore dimensionale necessario per rendere dimensionalmente
consistente luguaglianza. Poich una libbra forza viene definita come la forza che
accelera una massa unitaria di unaccelerazione pari a quella di gravit (32,174 [ft s
-2
]),
il valore necessario per rendere dimensionalmente consistente luguaglianza (11) pari
a g
c
= 32,174 [lb
m
ft lb
f
-1
s
-2
]. Questo fattore dimensionale viene spesso ritrovato nelle
relazioni che utilizzano le unit di misura del sistema anglosassone. Se le stesse
relazioni vengono usate con le unit di misura SI, deve ovviamente essere utilizzato un
valore g
c
= 1.
moltiplicando il valore in per il fattore si ottiene il valore in
ft 0,3048 m
ft
2
9,290 10
-2
m
2
ft
3
2,832 10
-2
m
3
lb
m
4,536 10
-1
kg
lb
mol
4,536 10
-1
kmol
Btu 1,055 kJ
Btu s
-1
1,055 kW
R 5/9 K
psf 4,788 10
-2
kPa
Tabella 1: fattori di conversione tra unit del Sistema Anglosassone e del SI.
Larea viene misurata in [ft
2
], mentre il volume in [ft
3
]. Analogamente, lunit di misura
della pressione [lb
f
ft
-2
] = [psf], mentre quella del lavoro (o del calore e dellenergia)
la British Thermal Unit, [Btu] e la potenza si misura in [Btu s
-1
].
Le relazioni tra le principali grandezze del SI e del Sistema Anglosassone sono riassunte
nella Tabella 1.
1.3.4 Altre unit di misura
Altre unit di misura molto usate sono:
per la lunghezza, pollici, [in];
per la superficie, pollici quadri, [in
2
];
per la pressione, atmosfere [atm]; torricelli (o mm di mercurio), [torr] = [mm
Hg
];
libbre forza per pollice quadro, [lb
f
in
-2
] = [psi]; metri di colonna dacqua, [m
H2O
];
per lenergia, calorie [cal];
per la temperatura, gradi celsius, [C], o fahrenheit, [F];
per il volume, galloni [gal]
12
, o litri [l];
12
Vi sono diverse definizioni del gallone; qui si riporta quella statunitense relativa ai liquidi.
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
13
per la potenza, cavalli [hp]
13
.
Le relazioni tra queste grandezze e le equivalenti del SI sono riassunte nella Tabella 2.
moltiplicando
14
il valore in per il fattore si ottiene il valore in
in 2,540 cm
in
2
6,452 cm
2
in
3
1,639 10
1
cm
2
cal 4,184 J
C C + 273,15 K
F 5/9(F - 32) C
atm 1,013 bar
torr = mm
Hg
1,333 10
-1
kPa
psi = lb
f
in
-2
6,893 10
-2
bar
m
H2O
9,806 10
-2
bar
gal 3,785 10
-3
m
3
l 10
-3
m
3
hp 7,460 10
2
W
Tabella 2: fattori di conversione tra unit di uso comune e del SI.
Oltre alla pressione assoluta (lunica utilizzata in termodinamica), nellambito
dellingegneria viene anche utilizzata la pressione relativa rispetto al valore
atmosferico, cos che P = P
rel
+ 1 [atm]. Nella notazione anglosassone lunit di misura
della pressione viene seguita dal suffisso g (per gauge) quando il valore riferito alla
pressione relativa (per esempio, 6 [psig]).
1.4 Trasformazioni spontanee e condizioni di equilibrio
Lesperienza mostra come tutti i sistemi, a seconda dei vincoli a cui sono sottoposti,
evolvono spontaneamente in una ben definita direzione. Per esempio, il calore fluisce
sempre spontaneamente da un corpo caldo a uno freddo e mai viceversa.
Cos come stato necessario introdurre il concetto di energia interna (una grandezza
non misurabile) per spiegare la possibilit di accumulare energia da parte di un sistema
in quiete, necessario introdurre il concetto di entropia (unaltra grandezza non
misurabile) per dare conto della direzionalit delle trasformazioni spontanee. Poich
lentropia fornisce informazioni sulla direzione verso cui evolve spontaneamente un
13
Vi sono diverse definizioni per il cavallo; qui si riporta quella statunitense relativa allacqua.
14
A parte ovviamente le relazioni per C e F, per cui si utilizzano le formule riportate.
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
14
sistema essa anche in grado di definire lo stato di equilibrio di un sistema come quello
stato da cui il sistema non ha alcuna tendenza ad evolvere spontaneamente.
Lentropia una grandezza estensiva funzione di stato definita in modo tale che, a
seguito di una trasformazione spontanea in un sistema isolato, essa pu solo aumentare.
Di conseguenza, le condizioni di equilibrio in un sistema isolato saranno caratterizzate
dal fatto che lentropia assume il valore massimo compatibile coi vincoli imposti al
sistema stesso.
Poich in condizioni di equilibrio lentropia di un sistema isolato massima, in una
trasformazione reversibile (che procede cio per stati di quasi equilibrio) la variazione
di entropia nulla.
Viceversa, per trasformazioni reversibili in sistemi chiusi la variazione di entropia
correlata unicamente al calore scambiato dalla relazione:
T
dQ
dS = (12)
Come detto in precedenza, gli stati di equilibrio possono essere descritti con un numero
limitato di variabili. Si visto che per un sistema monofase e monocomponente
sufficiente conoscere il valore di due variabili intensive per definirne in modo univoco
lo stato intensivo. Se invece si vuole definire lo stato estensivo di un sistema monofase
e monocomponente, necessario conoscere oltre al valore di due grandezze intensive
anche il valore del numero di moli (o della massa) presente nel sistema. Moltiplicando il
valore delle grandezze specifiche (intensive) per il numero di moli (o per la massa) si
risale al valore delle grandezze estensive. Appare evidente che, se noto il numero di
moli (o la massa), le stesse informazioni fornite dalla conoscenza di due grandezze
specifiche (intensive) possono essere desunte dalla conoscenza del valore delle stesse
grandezze estensive
15
. Per esempio, nulla cambia se si conosce il valore di u e v, oppure
quello di U, V e n, se non che nel secondo caso noto non solo lo stato intensivo del
sistema, ma anche quello estensivo:
estensivo ed intensivo stato : ) , , ( intensivo stato : ) , ( n V U v u (13)
Analogamente, si riscontra sperimentalmente che possibile descrivere completamente
lo stato estensivo di equilibrio di un sistema termodinamico
16
monofase ma
15
Per definizione, il valore della grandezza intensiva (per esempio il volume molare, v) viene
calcolato da quello della stessa grandezza estensiva (V) diviso il numero di moli (n): v = V / n.
Conoscere quindi due grandezze specifiche la stessa cosa che conoscere le due relative
grandezze estensive e il numero di moli.
16
Pi precisamente, lo stato di equilibrio stabile di un sistema isotropo.
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
15
multicomponente sulla base della conoscenza del valore dallenergia interna, del
volume e del numero di moli di ciascun composto presente nel sistema
17
.
Un sistema composto da pi fasi (sia monocomponente sia multicomponente) risulta
dallunione di pi sistemi monofase, per ciascuno dei quali valgono le considerazioni
fatte in precedenza. Il valore complessivo di una generica variabile estensiva
calcolabile semplicemente come somma delle variabili estensive relative a ciascuna
fase. Per esempio, il volume totale di un sistema composto da una fase liquida e da una
fase vapore dato dalla somma del volume della fase liquida e del volume della fase
vapore.
Poich lentropia una propriet intrinseca del sistema correlata alle sue variabili di
stato, deve essere possibile calcolarla sulla base dei valori dallenergia interna, del
volume e del numero di moli:
) , , ( n V U S S =
(14)
Inoltre, in condizioni di equilibrio in un sistema isolato il suo valore deve essere
massimo compatibilmente coi vincoli imposti al sistema stesso
18
, e quindi deve essere
nullo il suo differenziale primo (si ricordi che essendo lentropia una funzione di stato il
suo differenziale esatto):
=
=
|
|
.
|
\
|
+ |
.
|
\
|
+ |
.
|
\
|
N
i
i
n V U
i U V
dn
n
S
dV
V
S
dU
U
S
dS
i j
1
, ,
, ,
0
n n
(15)
Ponendo
19
:
17
Le leggi della termodinamica possono essere dedotte da alcuni postulati (o leggi) fondamentali.
In particolare, questa affermazione viene solitamente indicata come primo postulato nella
seguente forma: esistono particolari stati di un sistema isotropo, chiamati stati di equilibrio
stabile, che sono completamente caratterizzati dai valori dellenergia interna, U, del volume, V, e
del numero di moli di tutti i composti presenti, n.
18
Questo conduce al secondo postulato: esiste una funzione dei parametri estensivi di un sistema
(U, V e n) chiamata entropia, S, che definita per tutti gli stati di equilibrio e gode della seguente
propriet: i valori assunti da U, V e n in assenza di vincoli interni in un sistema isolato sono tali
da rendere massimo il valore di S.
19
Queste relazioni e quelle seguenti sono corrette grazie a quanto affermato dal terzo postulato:
lentropia di un sistema composto da pi sottosistemi data dalla somma dellentropia dei
singoli sottosistemi, cio S una grandezza estensiva; lentropia una funzione continua e
differenziabile monotonamente crescente con lenergia interna. Si veda lAppendice A per i
dettagli matematici.
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
16
T U
S
T
S
U
V V
1
, ,
= |
.
|
\
|
= |
.
|
\
|
n n
(16)
T
P
V
U
U
S
V
S
P
V
U
S V U S
= |
.
|
\
|
|
.
|
\
|
= |
.
|
\
|
= |
.
|
\
|
n n n n , , , ,
(17)
T n
U
U
S
n
S
n
U
i
n V S
i V
n V U
i
i
n V S
i
i j i j i j
=
|
|
.
|
\
|
|
.
|
\
|
=
|
|
.
|
\
|
=
|
|
.
|
\
|
, ,
,
, , , ,
n
(18)
si ricava:
=
= + =
N
i
i
i
dn
T
dV
T
P
dU
T
dS
1
0
1
(19)
da cui si possono facilmente dedurre le condizioni di equilibrio al trasferimento di
calore, di lavoro meccanico e di materia. Considerando un sistema isolato diviso in due
sottosistemi (A e B)
20
da una parete come illustrato in Figura 5, si assuma che la parete
sia impermeabile, rigida e diatermana.
Poich lentropia una variabile estensiva e sia il volume sia il numero di moli dei due
sottosistemi non pu cambiare (dn
A
= dn
B
= dV
A
= dV
B
= 0):
( ) 0
1 1
= + = + = + =
B
B
A
A
B A B A
dU
T
dU
T
dS dS S S d dS (20)
Essendo il sistema nel complesso isolato la sua energia interna non pu cambiare
21
e
quindi:
B A B A
dU dU U U U = = + = cost (21)
da cui segue:
0
1 1
= |
.
|
\
|
A
B A
dU
T T
(22)
20
Tutti i problemi della termodinamica sono sostanzialmente riconducibili a questa
schematizzazione.
21
Questo pu essere dedotta in maniera banale utilizzando il bilancio di energia discusso nel
capitolo seguente.
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
17
Figura 5: sistema isolato diviso in due sottosistemi da una parete.
La relazione precedente soddisfatta solo se T
A
=T
B
. La grandezza T assume quindi il
significato di temperatura e la relazione (22) richiede che in condizioni di equilibrio le
temperature dei sistemi a contatto con pareti diatermane siano uguali. In altri termini, la
condizione di equilibrio al trasferimento di calore che le temperature siano uguali.
In modo assolutamente analogo si ricavano le condizioni di equilibrio al trasferimento
di lavoro meccanico.
In questo caso si considera la parete impermeabile, diatermana e mobile e quindi, con
ragionamenti analoghi ai precedenti, si ricava che dn
A
= dn
B
= 0; dV
A
= - dV
B
; dU
A
= -
dU
B
. La condizione di massimo di entropia diviene quindi:
( )
0
1 1
1 1
= |
.
|
\
|
+ |
.
|
\
|
=
= + + + =
= + = + =
A
B A
A
B A
B
B
B
B
A
A
A
A
B A B A
dV
T
P
T
P
dU
T T
dV
T
P
dU
T
dV
T
P
dU
T
dS dS S S d dS
(23)
che soddisfatta (dovendo essere T
A
=T
B
) solo se P
A
=P
B
. La grandezza P assume quindi
il significato di pressione e la relazione (23) richiede che in condizioni di equilibrio le
pressioni dei sistemi a contatto con pareti mobili siano uguali. In altri termini, la
condizione di equilibrio al trasferimento di lavoro meccanico che le pressioni siano
uguali.
A B
PARETE RIGIDA,
ADIABATICA,
IMPERMEABILE
PARETE
DIVISORIA
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
18
Infine, si ricavano analogamente le condizioni di equilibrio al trasferimento di materia.
In questo caso si considera la parete permeabile, diatermana e mobile. Questo implica
B
i
A
i
dn dn = ; dV
A
= - dV
B
; dU
A
= - dU
B
e
( )
0
1 1
1 1
1
1 1
=
|
|
.
|
\
|
|
.
|
\
|
+ |
.
|
\
|
=
= + + + =
= + = + =
=
= =
N
i
A
i
B
B
i
A
A
i A
B A
A
B A
N
i
B
i
B
i B
B
B
B
N
i
A
i
A
i A
A
A
A
B A B A
dn
T T
dV
T
P
T
P
dU
T T
dn
T
dV
T
P
dU
T
dn
T
dV
T
P
dU
T
dS dS S S d dS
(24)
che soddisfatta (dovendo essere T
A
=T
B
e P
A
=P
B
) solo se
B
i
A
i
= . La grandezza
i
,
il cui senso fisico meno intuitivo rispetto a quello di temperatura e pressione, prende il
nome di potenziale chimico
22
. La relazione (24) richiede che in condizioni di equilibrio
il potenziale chimico di ciascuna specie presente in sistemi separati da pareti
permeabili
23
siano uguali. In altri termini, la condizione di equilibrio al trasferimento di
materia che i potenziali chimici di ciascuna specie nei diversi sottosistemi siano
uguali.
22
Il potenziale chimico del composto i-esimo pu essere visto come la variazione di energia
interna di un sistema a seguito dellaggiunta di una mole del composto i-esimo, aggiunta fatta
mantenendo costanti lentropia e il volume del sistema, oltre che il numero di moli di tutte le altre
specie.
23
Un tipico esempio di sottosistemi separati da una parete permeabile rappresentato da una fase
liquida e una fase vapore a contatto.
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
19
2 Bilanci in sistemi monocomponente
La scrittura dei bilanci relativi a una grandezza estensiva riveste un ruolo assolutamente
centrale nellingegneria in generale e in quella chimica in particolare. Si tratta di uno
strumento di indagine estremamente potente che consente, tra laltro, di dedurre i noti
principi della termodinamica come casi particolari di bilanci pi generali.
Al termine dello studio di questo capitolo lo studente dovrebbe conoscere:
come formulare in termini generali il bilancio di una grandezza estensiva;
il bilancio di materia in termini molari e massici, a stazionario e in transitorio;
il bilancio di energia in termini molari e massici, a stazionario e in transitorio;
il lavoro di pompaggio;
lentalpia;
il primo principio della termodinamica nelle sue diverse formulazioni;
le definizioni di capacit termica;
come calcolare il calore e il lavoro scambiati in trasformazioni semplici;
gli stati di riferimento per il calcolo di entalpia e energia interna;
il bilancio di entropia;
il secondo principio della termodinamica nelle sue varie formulazioni;
il gas perfetto e la sua equazione di stato;
come calcolare le differenze di entalpia, entropia ed energia interna specifica tra due
stati per un gas perfetto;
la relazione tra il calore specifico a pressione e a volume costante per un gas perfetto;
come calcolare le grandezze termodinamiche ed il calore e lavoro scambiati da un gas
perfetto per trasformazioni semplici.
2.1 Formulazione generale di un bilancio
Un bilancio la traduzione in termini matematici dellaffermazione che, per una data
grandezza estensiva (per esempio il numero di moli) la somma dei flussi netti entranti in un
sistema e della quantit netta prodotta nellunit di tempo allinterno del sistema stesso
deve essere uguale alla variazione netta nel tempo della quantit presente allinterno del
sistema stesso.
Col termine flussi netti entranti nel sistema si intende la differenza tra i flussi entranti e
quelli uscenti; con quantit netta prodotta la differenza tra la quantit prodotta e quella
consumata nellunit di tempo; con variazione netta nel tempo la differenza tra laumento e
la diminuzione della quantit presente allinterno del sistema stesso, sempre nellunit di
tempo. In tutti i casi quindi, un valore positivo indica il prevalere del termine entrante
rispetto a quello uscente; del termine di produzione rispetto a quello di consumo; del
termine di accumulo rispetto a quello di depauperamento. Per esempio, una variazione netta
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
20
pari a -10 [kg s
-1
] indica che la massa presente nel sistema diminuisce di 10 [kg] ogni
secondo.
Questa affermazione applicata alla materia, allenergia e allentropia (pur di computare
correttamente i vari termini) non mai stata smentita da alcuna evidenza sperimentale e la
si ritiene quindi di validit generale (si tratta, in altri termini, di una legge). Inoltre,
invalso luso di separare, nella scrittura delle equazioni di bilancio, i flussi associati alle
portate massiche che attraversano la superficie di controllo dagli altri flussi e di considerare
separatamente i contributi associati alle portate massiche entranti da quelli associati alle
portate massiche uscenti.
La formulazione generale di una equazione di bilancio risulta quindi essere:
accumulo = in
mat
- out
mat
+ flussi in + generazione (25)
dove tutti i termini hanno dimensioni pari allunit di misura della grandezza estensiva
sottoposta a bilancio diviso un tempo. Per esempio, i termini del bilancio di materia
possono essere espressi in [kg s
-1
], mentre quelli del bilancio di energia in [J s
-1
] = [W].
Il termine accumulo positivo se prevale laumento allinterno del sistema rispetto alla
diminuzione; i flussi associati alle portate materiali che entrano, in
mat
, o escono, out
mat
, dal
sistema sono entrambi positivi grazie al segno meno posto davanti al termine uscente; il
termine flussi in tiene conto dei flussi che attraversano la superficie di controllo senza
essere associati a flussi materiali e risulta positivo se prevalgono i contributi in ingresso,
mentre negativo se prevalgono quelli in uscita; infine, il termine di generazione positivo
se prevale la quantit prodotta su quella consumata nellunit di tempo e negativo
viceversa.
2.2 Bilancio di materia
Considerando come grandezza estensiva la quantit di materia la equazione generale (25)
diventa:
OUT IN
n n
dt
dn
& & = (26)
dove n [mol] il numero totale di moli presenti nel sistema, mentre
IN
n& e
OUT
n& [mol s
-1
]
rappresentano le portate molari entranti ed uscenti dal sistema, rispettivamente. Si noti che,
per un sistema monocomponente, il termine di generazione della materia (escludendo
situazioni non contemplate nella termodinamica dellingegneria chimica, quali le reazioni
nucleari) sempre nullo.
Per i sistemi monocomponente esiste poi una relazione biunivoca tra la massa e il numero
di moli attraverso il peso molecolare M (costante) del composto:
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
21
] [ ] [ ] [
] [ ] [ ] [
1 1 1
1
=
=
kmol kg M s kmol n s kg m
kmol kg M kmol n kg m
& &
(27)
Questo consente di esprimere lequazione di bilancio della quantit di materia nei termini
equivalenti di massa:
OUT IN
m m
dt
dm
& & = (28)
dove m [kg] la massa totale presente nel sistema, mentre
IN
m& e
OUT
m& [kg s
-1
]
rappresentano le portate massiche entranti ed uscenti dal sistema, rispettivamente.
2.2.1 Condizioni stazionarie
Si definiscono stazionarie quelle condizioni per cui il termine di accumulo risulta nullo, e
quindi la quantit della grandezza estensiva sottoposta a bilancio presente nel sistema non
varia nel tempo.
Le equazioni riportate sopra diventano quindi in condizioni stazionarie:
OUT IN
n n & & = (29)
OUT IN
m m & & = (30)
sancendo semplicemente che ci che entra pari a ci che esce.
Si noti che questo vero in termini di portate massiche e molari, ma non necessariamente
in termini di portate volumetriche. Infatti, la portata volumetrica, F [m
3
s
-1
], legata alla
portata massica dalla densit massica, [kg m
-3
], e quindi la relazione (30) diventa:
OUT OUT IN IN
F F = (31)
Solo nel caso in cui
OUT IN
= anche la portata volumetrica entrante uguale a quella
uscente. Questo solitamente il caso, come verr discusso pi avanti, dei liquidi la cui
densit massica pu spesso essere considerata costante.
2.2.2 Applicazione
Si calcoli il tempo necessario a riempire di acqua una vasca da 0.2 [m
3
] utilizzando un
rubinetto che eroga una portata costante di 0.5 [kg s
-1
] se presente una perdita costante di
0.01 [kg s
-1
].
La massa di acqua presente nella vasca piena, m, si pu calcolare come:
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
22
] [ 200 ] [ 1000 ] [ 2 . 0
3 3
kg m kg m V m = = =
(32)
in cui si considerata la densit massica dellacqua pari a 1000 [kg m
-3
].
La relazione (26) in questo caso diventa:
t dt dm
dt
dm
t
49 . 0 200 49 . 0 01 . 0 5 . 0
0
200
0
= = =
(33)
Il tempo necessario quindi pari a 200 / 0.49 = 408 [s].
2.3 Bilancio di energia
Considerando come grandezza estensiva lenergia, lequazione generale (25) diventa:
W Q E E
dt
dE
OUT IN
& & & &
+ + = (34)
dove E [J] lenergia totale del sistema, mentre
IN
E
&
ed
OUT
E
&
[W] rappresentano i flussi di
energia associati alle portate materiali entranti ed uscenti dal sistema, rispettivamente. Q
&
e
W
&
[W] sono invece le potenze termiche e meccaniche nette che attraversano la superficie
di controllo del sistema. Trattandosi di termini netti, rappresentano la differenza tra i flussi
entranti e quelli uscenti; di conseguenza, un valore positivo implica una potenza entrante
nel sistema, mentre un valore negativo una potenza uscente.
I contributi tipici presenti nel termine di potenza meccanica sono normalmente quelli
trasmessi attraverso la rotazione di un albero (pompe, compressori, turbine, agitatori) e
quelli di espansione legati alla variazione di volume del sistema (sistemi cilindro - pistone).
Possono per essere presenti anche altri contributi, solitamente esprimibili come prodotto di
una grandezza intensiva per la derivata temporale di una grandezza estensiva:
dt
db
a W
ab
=
&
(35)
Un esempio la potenza elettrica se a il potenziale elettrico e b la quantit di carica. Si
noti che anche il lavoro di espansione rientra in questa tipologia ponendo a pari alla
pressione e b uguale al volume.
Il termine di produzione dellenergia (escludendo situazioni non contemplate nella
termodinamica dellingegneria chimica, quali le reazioni nucleari) sempre nullo.
Lenergia totale del sistema, per quanto solitamente di interesse per lingegneria chimica,
composta essenzialmente da tre contributi. Questi sono lenergia cinetica, E
k
, quella
potenziale, E
p
, e quella interna, U:
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
23
|
|
.
|
\
|
+ + =
|
|
.
|
\
|
+ + = + + = u Mgz
v
M n u gz
v
m U E E E
p k
2
2
2 2
(36)
dove, come detto in precedenza, u [J mol
-1
] indica lenergia interna molare e u [J kg
-1
]
quella massica. Nei problemi tipici dellingegneria solitamente tutti i tre termini possono
essere importanti, mentre in quelli dellingegneria chimica risulta di solito prevalente il
termine di energia interna. In questo caso, le relazioni precedenti possono essere
approssimate dalle seguenti:
nu u m U E = = (37)
Analogamente, associata ai flussi materiali entranti ed uscenti dal sistema vi una certa
quantit di energia cinetica, legata alla velocit del flusso, potenziale, legata alla quota a cui
il flusso materiale attraversa la superficie di controllo, e interna, legata alle condizioni
termodinamiche del flusso (temperatura e pressione). Inoltre, un fluido in moto esercita un
lavoro sulla porzione di fluido davanti ad esso (rispetto alla direzione del moto) per
vincerne la pressione e, analogamente, riceve un lavoro dal fluido dietro di esso. In altri
termini, lambiente compie un lavoro contro la pressione prevalente nella sezione di
ingresso per spingere il fluido nel sistema e il sistema compie un lavoro contro la
pressione prevalente nella sezione di uscita per spingere il fluido nellambiente. Questo
particolare tipo di lavoro necessario per il moto di un fluido prende comunemente il nome
di lavoro di pompaggio.
Con riferimento alla Figura 6, per quantificare questo lavoro si considerino due cilindri di
fluido, contenenti entrambi lunit di massa di fluido: il primo nelle condizioni di ingresso
della portata massica, IN, il secondo nelle condizioni di uscita, OUT.
Il volume di entrambi i cilindri quindi pari al volume massico, v , nelle diverse condizioni
IN,
IN
v , e OUT,
OUT
v . La lunghezza (per unit di massa) del cilindro 1 quindi pari a
IN IN
A v L /
1
= (essendo A
IN
la superficie della sezione di ingresso del fluido) e la forza che
esercita sul sistema uguale a
IN IN
A P F =
1
(dove P
IN
la pressione nella sezione di
ingresso). Il lavoro fatto dallambiente sul sistema per introdurre il cilindro 1 nel sistema
stesso quindi pari al prodotto della forza per lo spostamento:
IN IN
IN
IN
IN IN IN
v P
A
v
A P W
= =
(38)
Poich il cilindro contiene lunit di massa di fluido, si tratta di lavoro per unit di massa
fluente nel sistema, [J kg
-1
]. La potenza entrante nel sistema associata a una data portata
massica
IN
m& sar quindi pari a:
IN IN IN IN
v P m W &
&
= (39)
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
24
Analogamente, considerando questa volta il cilindro 2, la potenza associata al flusso
materiale uscente dal sistema,
OUT
m& , sar quindi pari a:
OUT OUT OUT OUT
v P m W &
&
= (40)
I flussi di energia associati ai flussi di materia si possono quindi esprimere in generale
come:
|
|
.
|
\
|
+ + + =
|
|
.
|
\
|
+ + + = Pv u Mgz
v
M n v P u gz
v
m E
2
2
2 2
& &
&
(41)
Figura 6: lavoro di pompaggio.
Q
OUT
IN
SUPERFICIE DI
CONTROLLO
1 kg NELLE
CONDIZIONI
OUT (cilindro 2)
1 kg NELLE
CONDIZIONI
IN (cilindro 1)
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
25
Nei termini di energia associata ai flussi materiali il termine di energia interna risulta
sempre associato al lavoro di pompaggio. Risulta quindi utile definire una nuova funzione
termodinamica, chiamata entalpia, H, e definita come:
PV U H + = (42)
Analogamente allenergia interna, lentalpia una grandezza estensiva con le dimensioni di
unenergia, [J]. Le relative grandezze specifiche sono ovviamente:
v P u h
Pv u h
+ =
+ =
(43)
La relazione (41) assume quindi la forma pi compatta:
|
|
.
|
\
|
+ + =
|
|
.
|
\
|
+ + = h Mgz
v
M n h gz
v
m E
2
2
2 2
& &
&
(44)
e il bilancio di energia diventa:
W Q h gz
v
m
h gz
v
m u gz
v
m
dt
d
OUT OUT
OUT
OUT
IN IN
IN
IN
& &
&
&
+ +
|
|
.
|
\
|
+ +
+
|
|
.
|
\
|
+ + =
(
(
|
|
.
|
\
|
+ +
2
2
2 2
(45)
Si noti che nel termine di accumulo compare lenergia interna, mentre in quelli associati
alle portate materiali lentalpia.
Analogamente a quanto detto in precedenza, nei problemi tipici dellingegneria tutti i
termini presenti nella relazione (44) possono essere importanti, mentre in quelli
dellingegneria chimica risulta di solito prevalente il termine di entalpia. In questo caso, i
flussi di energia possono essere approssimati come:
h n h m E & &
&
=
(46)
e il bilancio di energia approssimato diventa:
( )
W Q h m h m
dt
u m d
dt
dU
OUT OUT IN IN
& &
& & + + = =
(47)
2.3.1 Condizioni stazionarie
In condizioni stazionarie il bilancio di energia diventa:
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
26
( ) ( )
0
2
2 2
, , , ,
= + +
|
|
.
|
\
|
+ +
|
|
.
|
\
|
+ +
= + + + + + +
W Q h gz
v
m h gz
v
m
W Q H E E H E E
OUT OUT
OUT
OUT IN IN
IN
IN
OUT OUT p OUT k IN IN p IN k
& &
& &
& & & & & & & &
(48)
Sempre in condizioni stazionarie il bilancio di materia (30) richiede che le portate massiche
entranti e uscenti siano uguali:
m m m
OUT IN
& & & = = (49)
e quindi il bilancio di energia in condizioni stazionarie assume la forma:
0
2
2
2 2
2 2
= + +
|
|
.
|
\
|
+ +
|
|
.
|
\
|
+ + =
= + +
|
|
.
|
\
|
+ +
|
|
.
|
\
|
+ +
W Q h gz
v
h gz
v
m
W
m
Q
h gz
v
h gz
v
OUT OUT
OUT
IN IN
IN
OUT OUT
OUT
IN IN
IN
&
&
&
&
(50)
Tutti i termini in questa relazione hanno dimensioni di energia massica, [J kg
-1
], e i due
ultimi termini ( Q
e W
) hanno quindi il significato di quantit di calore e di lavoro fornita
per kg di materia fluente nel sistema.
Nelle condizioni in cui i termini di energia cinetica e potenziale sono trascurabili rispetto a
quello entalpico il bilancio assume la forma approssimata:
0
= + + W Q h h
OUT IN
(51)
che prende il nome di bilancio entalpico.
Da questo bilancio si deduce facilmente che il flusso attraverso un sistema che non scambia
n calore n lavoro con lambiente (sempre nellipotesi di trascurare i contributi cinetici e
potenziali) isoentalpico. In altri termini, come mostrato nella Figura 7 per il classico
esempio della valvola di laminazione isoentalpica, lentalpia specifica della corrente
entrante uguale a quella della corrente uscente
2.3.2 Primo principio della termodinamica
Il primo principio della termodinamica pu essere enunciato in molti modi equivalenti, tutti
deducibili dalla forma generale del bilancio di energia.
Uno dei pi semplici ed intuitivi il seguente:
Lenergia pu assumere diverse forme, ma la quantit totale di energia delluniverso
costante
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
27
Luniverso un sistema isolato, che cio non scambia n materia n energia. Il bilancio di
energia (45) applicato a un sistema isolato assume la forma:
0
2
2
=
(
(
|
|
.
|
\
|
+ + u gz
v
m
dt
d
(52)
da cui, integrando, deriva:
costante = + + U E E
p k
(53)
che la traduzione in termini matematici dellaffermazione precedente.
Unaltra formulazione molto comune del primo principio quella che deriva dagli
esperimenti di Joule ed afferma che:
La variazione dellenergia interna di un sistema chiuso pari alla somma del calore e del
lavoro netti entranti nel sistema
Figura 7: valvola di laminazione isoentalpica. In queste condizioni
OUT IN
h
= .
Negli esperimenti di Joule si analizza un sistema chiuso e fermo, in cui cio non vi sono
flussi di materia in ingresso o uscita e la variazione di energia cinetica e potenziale del
sistema nulla. In queste condizioni il bilancio di energia (45) diventa:
IN
OUT
SUPERFICIE DI
CONTROLLO
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
28
( )
W Q
dt
u m d
& &
+ =
(54)
da cui:
dW dQ dt W dt Q dU + = + =
& &
(55)
che la nota traduzione in termini matematici dellaffermazione precedente. Se si considera
poi solo un lavoro di espansione reversibile, introducendo la relazione (4) si ottiene:
PdV dQ dU = (56)
Una terza formulazione del primo principio, anchessa derivante dagli esperimenti di Joule,
afferma che:
In una trasformazione ciclica la somma del calore e del lavoro netto uguale a zero
Una trasformazione si dice ciclica se lo stato iniziale coincide con quello finale. Di
conseguenza, in una trasformazione ciclica non vi alcuna variazione delle funzioni di
stato. In questo caso la relazione (55), integrata su tutta la trasformazione ciclica, diventa:
( ) W Q U dW dQ dU + = + =
(57)
Poich lenergia interna una funzione di stato la sua variazione in una trasformazione
ciclica nulla e la relazione precedente diventa:
( ) 0 0 = + = +
W Q dW dQ (58)
che la traduzione in termini matematici dellaffermazione precedente.
Questa relazione implica anche che non possibile costruire una macchina che funzioni
secondo una trasformazione ciclica in grado di fornire lavoro in modo continuo senza
assorbire calore dallambiente. In altri termini, non possibile costruire una macchina in
moto perpetuo di primo tipo.
2.3.2.1 Trasformazioni semplici
Se si considera una trasformazione isocora la relazione (56) diventa:
dQ dU = (59)
Integrando questa relazione tra uno stato iniziale e uno finale si ottiene:
Q U =
(60)
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
29
che mostra come il calore scambiato in una trasformazione isocora (ovviamente, sotto le
condizioni di sistema chiuso, fermo e di lavoro di espansione reversibile per cui valida la
relazione (56)) eguaglia la variazione di energia interna.
Se invece si considera una trasformazione isobara la relazione (56) diventa:
dH PV U d PV d dU PdV dU dQ = + = + = + = ) ( ) (
(61)
e quindi:
Q H =
(62)
che mostra come il calore scambiato in una trasformazione isobara (sempre sotto le
condizioni di validit della relazione (56)) eguaglia la variazione di entalpia.
La capacit termica (o calore specifico) misura la reazione di un sistema allingresso di
calore in termini di variazione di temperatura:
dT
dQ
C = (63)
Una tale definizione di capacit termica ha lo svantaggio di dipendere dalla trasformazione
considerata in quanto il calore non una funzione di stato. Si utilizzano allora due diverse
definizioni che, coinvolgendo solo funzioni di stato, non dipendono da una particolare
trasformazione:
P
P
P
P
P
P
T
h
c
T
h
c
T
H
C |
.
|
\
|
=
|
|
.
|
\
|
= |
.
|
\
|
= ;
(64)
V
V
V
V
V
V
T
u
c
T
u
c
T
U
C |
.
|
\
|
= |
.
|
\
|
= |
.
|
\
|
= ;
(65)
La prima viene chiamata capacit termica a pressione costante, mentre la seconda a volume
costante.
Queste quantit possono essere misurate effettuando delle trasformazioni reversibili in
sistemi chiusi per cui il calore scambiato a pressione costante coincide con la variazione di
entalpia, mentre quello scambiato a volume costante con la variazione di energia interna.
Sempre sotto le condizioni per cui valida la relazione (56), si possono ottenere delle
relazioni per calcolare la variazione delle funzioni di stato e il calore scambiato in
trasformazioni semplici.
Lenergia interna massica una grandezza intensiva e quindi, per un sistema monofase e
monocomponente, funzione di sole altre due variabili intensive. In particolare la si pu
considerare funzione della temperatura e del volume specifico: ( ) v T u u , = . Trattandosi di
una funzione di stato, il suo differenziale esatto pu essere espresso come:
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
30
v d
v d
u
dT c v d
v d
u
dT
dT
u
u d
T
V
T v
|
.
|
\
|
+ = |
.
|
\
|
+ |
.
|
\
|
= (66)
Fisicamente questo significa che la variazione di energia interna specifica pu essere
calcolata come somma di due contributi: la variazione di energia interna specifica dovuta
alla variazione unitaria di temperatura (pari a
V
c ) per la variazione infinitesima di
temperatura e la variazione di energia interna specifica dovuta alla variazione unitaria di
volume specifico (pari a ( )
T
v d u / ) per la variazione infinitesima di volume specifico.
Per trasformazioni isocore in sistemi chiusi anche il volume massico rimane costante
24
e
quindi sotto le condizioni di validit della relazione (56) la relazione precedente diventa:
dT c u d
V v
= (67)
che introdotta nella relazione (60) fornisce un mezzo per il calcolo della variazione di
energia interna e del calore scambiato in una trasformazione isocora:
( )
= = = =
v T
v T
V
v T
v T
v T
v T
dT c m u d m u m d U Q
,
,
,
,
,
,
2
1
2
1
2
1
(68)
Si noti che la relazione precedente pu essere utilizzata per calcolare la variazione di
energia interna anche per una trasformazione non isocora, ma tale per cui il volume massico
iniziale e finale coincidono. Questa una diretta conseguenza del fatto che lenergia interna
una funzione di stato e quindi la sua variazione dipende solo dallo stato iniziale e finale.
Ovviamente, per questa trasformazione non isocora il calore scambiato, che non una
funzione di stato, non sar uguale alla variazione di energia interna.
Analogamente, si pu considerare lentalpia massica funzione della temperatura e della
pressione: ( ) P T h h ,
= . Trattandosi di una funzione di stato, il suo differenziale esatto pu
essere espresso come:
dP
dP
h
dT c dP
dP
h
dT
dT
h
h d
T
P
T P
|
|
.
|
\
|
+ =
|
|
.
|
\
|
+
|
|
.
|
\
|
=
(69)
dal significato analogo alla (66).
Per trasformazioni isobare in sistemi chiusi la relazione precedente diventa:
dT c h d
P P
= (70)
24
In quanto essendo costante sia il volume (traformazione isocora) sia la massa (sistema chiuso)
costante anche il volume massico ( m V v / = ).
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
31
che introdotta nella relazione (62) fornisce un mezzo per il calcolo della variazione di
entalpia e del calore scambiato in una trasformazione isobara:
( )
= = = =
P T
P T
P
P T
P T
P T
P T
dT c m h d m h m d H Q
,
,
,
,
,
,
2
1
2
1
2
1
(71)
Similmente al caso delle trasformazioni isocore, la relazione precedente pu essere
utilizzata per calcolare la variazione di entalpia anche per una trasformazione non isobara,
ma tale per cui la pressione iniziale e finale coincidono in quanto anche lentalpia una
funzione di stato. Ovviamente, per questa trasformazione non isobara il calore scambiato,
che non una funzione di stato, non sar uguale alla variazione di entalpia.
2.3.2.2 Stati di riferimento
Le relazioni sviluppate nella precedente sezione consentono di calcolare le variazioni di
energia interna e di entalpia. Risulta perci necessario definire uno stato di riferimento dove
si pone uguale a zero il valore di una di queste due funzioni.
Comunemente si sceglie un valore di temperatura, T
r
, dove si pone uguale a zero il valore
dellentalpia per un dato valore di pressione, P:
( ) ( )
= + =
T
T
P
T
T
P r
r r
dT c dT c P T h P T h ,
(72)
Per sistemi monocomponente, la scelta dello stato di riferimento completamente
arbitraria, ma deve essere mantenuta uguale per tutti i calcoli che vengono svolti. In
particolare, il valore dellenergia interna deve essere calcolato come:
( ) ( )
( ) ( ) ( )
+ = + =
= + =
T
T
V r
T
T
V r r
T
T
V r
r r
r
dT c P T v P dT c P T v P P T h
dT c P T u P T u
, , ,
, ,
(73)
Poich la scelta dello stato di riferimento arbitraria, essa non pu influenzare i risultati dei
conti. Di conseguenza, tutti i valori legati allo stato di riferimento devono elidersi nelle
formule risolutive del problema.
2.3.3 Applicazione
Un compressore aspira un gas a 25 [C] e 1 [bar] con una velocit trascurabile rispetto a
quella di scarico fornendogli una quantit di lavoro pari a 240 [kJ kg
-1
]. A valle del
compressore il gas scambia calore con un fluido ausiliario e viene poi scaricato a 25 [C], 1
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
32
[bar] e 300 [m s
-1
]. Si richiede di calcolare il calore scambiato col fluido ausiliario in
condizioni stazionarie.
Il problema pu essere schematizzato come riportato in Figura 8. Il bilancio di energia in
questo caso risulta essere:
( ) 0
2
2
= + +
|
|
.
|
\
|
+ + + W Q h gz
v
h gz
OUT OUT
OUT
IN IN
(74)
I termini di energia potenziale risultano trascurabili rispetto a quelli di energia cinetica a
causa dellalta variazione di velocit. Infatti 45000 2 / 300 2 /
2 2
= =
OUT
v [m
2
s
-2
]
dellordine di 10
4
, mentre anche un dislivello di 100 [m] implicherebbe un termine di
energia potenziale pari a ( ) 980 100 8 , 9 = =
OUT IN
z z g [m
2
s
-2
], dellordine quindi di 10
3
.
Figura 8: schematizzazione del problema.
Inoltre, lo stato della corrente in ingresso (25 [C] e 1 [bar]) uguale a quello della corrente
in uscita e quindi i valori di tutte le funzioni di stato sono uguali nelle due correnti. In
particolare, la variazione di entalpia specifica risulta nulla:
OUT IN
h h
= .
Si ottiene quindi:
IN
OUT
Q
&
W
&
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
33
] kg [J 195000 240000
2
300
1
2 2
= = W
v
Q
OUT
(75)
Essendo il risultato negativo, significa che si tratta di calore uscente dal sistema e quindi
che il gas viene raffreddato nello scambiatore di calore.
2.4 Bilancio di entropia
Considerando come grandezza estensiva lentropia la equazione generale (25) diventa:
S OUT IN
R
T
Q
S S
dt
dS
&
&
& &
+ + = (76)
dove S [J K
-1
] lentropia totale del sistema, mentre
IN
S
&
e
OUT
S
&
[W K
-1
] rappresentano i
flussi di entropia associati alle portate materiali entranti ed uscenti dal sistema,
rispettivamente. Come noto, solo il flusso di calore contribuisce alla variazione di
entropia del sistema con una velocit pari a T Q/
&
[W K
-1
], mentre la potenza meccanica
che attraversa la superficie di controllo non varia lentropia del sistema. Se la
trasformazione non reversibile, si ha poi anche un termine di generazione di entropia,
S
R
&
,
che per definizione positivo o nullo (essendo nullo solo in condizioni di equilibrio, e
quindi anche per trasformazioni reversibili).
Una prima conseguenza evidente dal bilancio di entropia che in un sistema chiuso
adiabatico la variazione di entropia nulla per trasformazioni reversibili. In altri termini, in
un sistema chiuso una trasformazione adiabatica reversibile anche isoentropica.
2.4.1 Condizioni stazionarie
In condizioni stazionarie il bilancio di entropia diventa:
0 = + +
S OUT IN
R
T
Q
S S
&
&
& &
(77)
In condizioni stazionarie il bilancio di materia (30) richiede che le portate massiche entranti
e uscenti siano uguali e quindi il bilancio di entropia in condizioni stazionarie assume la
forma:
0
= + + = + +
S OUT IN
S
OUT IN
R
T
Q
S S
m
R
m T
Q
S S
&
&
&
&
(78)
Tutti i termini in questa relazione hanno dimensioni di entropia massica, [J K
-1
kg
-1
], e
lultimo termine,
S
R
, ha quindi il significato di entropia generata per kg di materia fluente
nel sistema.
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
34
In condizioni adiabatiche e reversibili il bilancio si semplifica ulteriormente:
OUT IN
S S
= (79)
Da questo bilancio si deduce che il flusso in condizioni reversibili attraverso un sistema che
non scambia calore con lambiente isoentropico. In altri termini, come mostrato nella
Figura 9 per il classico esempio del compressore, lentropia specifica della corrente entrante
uguale a quella della corrente uscente.
Figura 9: flusso attraverso un compressore. In queste condizioni
OUT IN
S
= .
Anche per un sistema aperto in condizioni stazionarie, una trasformazione adiabatica
reversibile isoentropica.
2.4.2 Secondo principio della termodinamica
Il secondo principio della termodinamica pu essere enunciato in molti modi equivalenti,
tutti deducibili dalla forma generale del bilancio di entropia.
Uno dei pi semplici ed intuitivi il seguente:
Lentropia delluniverso pu solo aumentare.
IN
OUT
SUPERFICIE DI
CONTROLLO
W
&
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
35
Luniverso un sistema isolato, che cio non scambia n materia n energia. Il bilancio di
entropia (76) applicato a un sistema isolato assume la forma:
0 =
S
R
dt
dS
&
(80)
dove luguaglianza vale solo in condizioni di equilibrio, cio in assenza di gradienti di
temperatura, pressione e potenziale chimico. Questa relazione la traduzione in termini
matematici dellaffermazione precedente.
Unaltra formulazione, detta di Kelvin - Planck
25
, la seguente:
Non possibile costruire una macchina che funzioni secondo una trasformazione ciclica
il cui solo effetto sia quello di prelevare calore dallambiente e fornire allambiente una
equivalente quantit di lavoro
Figura 10. Macchina termica.
25
Max Karl Ernest Ludwing Planck, 1858 - 1927, fisico tedesco, premio Nobel nel 1918.
SERBATOIO DI
CALORE
CALDO
SERBATOIO DI
CALORE
FREDDO
MACCHINA
TERMICA
Q
H
Q
C
W
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
36
In altri termini, non possibile costruire una macchina in moto perpetuo di secondo tipo,
cio quella schematizzata in Figura 10 con Q
C
= 0.
Applicando il bilancio di entropia al serbatoio di calore
26
che si trova alla temperatura
maggiore, T
H
, si ottiene
H
H H
T
Q
dt
dS
&
= (81)
dove col pedice H si indicato il serbatoio di calore caldo. Il termine di generazione
nullo in quanto, poich la temperatura del serbatoio non cambia, leffetto di uno scambio
termico sul serbatoio lo stesso qualsiasi sia la temperatura del sistema con cui scambia
calore, cio sia che lo scambio termico sia reversibile o irreversibile.
Integrando questa espressione su di un intero ciclo
27
si ottiene:
H
H
H H
H
H
T
Q
S dt Q
T
dS = =
&
1
(82)
Analogamente, per il serbatoio di calore freddo (indicato col pedice C), si ottiene:
Tc
Q
S
C
C
= (83)
La variazione totale di entropia al termine del ciclo sar la somma di quella dei due serbatoi
e della macchina termica. Questultima per nulla in quanto, per definizione, la machina
termica al termine del ciclo ritorna nello stato iniziale e lentropia una funzione di stato.
Ne consegue quindi che:
Tc
Q
T
Q
S
C
H
H
+ =
(84)
Il bilancio di energia (nella forma del primo principio) applicato alla macchina termica su
di un intero ciclo rappresentato dalla equazione (58) che in questo caso diventa:
( ) 0 = + + W Q Q
C H
(85)
dove il segno meno tiene conto del fatto che il calore entrante nella macchina termica ha
segno opposto rispetto a quello entrante nei serbatoi di calore. Introducendo la relazione
(84) si ottiene:
26
Un serbatoio di calore un sistema chiuso isotermo e uniforme in grado di scambiare calore senza
variare la propria temperatura.
27
In questo caso la trasformazione ciclica ovviamente riferita al fluido utilizzato dalla macchina
termica.
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
37
|
|
.
|
\
|
+ = +
|
|
.
|
\
|
= + =
C
H
C H C
C
C
H C H
T
T
Q S T Q
T
Q
S T Q Q W 1 (86)
Il lavoro viene prodotto dalla macchina termica e quindi negativo. Coerentemente,
lultimo termine tra parentesi negativo. Viceversa, il termine S positivo o nullo e pu
quindi solo ridurre il lavoro prodotto se la trasformazione non reversibile.
Se Q
C
= 0 il lavoro non pu che essere positivo (cio entrante nella macchina termica) o
nullo: la relazione (86) la traduzione in termini matematici dellaffermazione precedente.
Infine, il secondo principio pu anche essere formulato come segue (formulazione di
Clausius
28
):
Non possibile costruire una macchina che funzioni secondo una trasformazione ciclica
il cui solo effetto sia quello di prelevare calore da una sorgente pi fredda e cederlo a una
sorgente pi calda.
In altri termini, il calore passa spontaneamente dai corpi caldi a quelli freddi.
Infatti, considerando due serbatoi di calore a contatto attraverso una parete diatermana, il
bilancio di entropia per ciascun serbatoio dato dalle relazioni (83) e (84), rispettivamente,
che forniscono la variazione di entropia per ciascun serbatoio al termine di una
trasformazione ciclica. La variazione totale di entropia, sempre al termine della
trasformazione ciclica, sar quindi:
0
|
|
.
|
\
|
= +
= + =
C H
C H
C
C
H
C C
H
H
T T
T T
Q
Tc
Q
T
Q
Tc
Q
T
Q
S (87)
In questa relazione si considerato che il calore uscente da un serbatoio uguale a quello
entrante nellaltro, e quindi uguale in valore assoluto ma di segno opposto. Poich il
termine tra parentesi positivo, affinch la variazione di entropia totale sia maggiore di
zero (cio che il trasporto di calore sia spontaneo) necessario che Q
C
sia positivo, e quindi
che si tratti di calore entrante nel serbatoio freddo. La relazione (87) quindi la
traduzione in termini matematici dellaffermazione precedente.
2.4.3 Combinazione del primo e del secondo principio
Considerando un sistema chiuso e monocomponente in cui avvenga una trasformazione
reversibile il bilancio di entropia (76) diventa:
28
Rudolf Clausius, 1822 1888, fisico tedesco.
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
38
T
Q
dt
dS
&
= (88)
oppure
TdS dQ
T
dQ
T
dt Q
dS = = =
&
(89)
Introducendo il bilancio di energia (56) valido per un sistema omogeneo chiuso e per
trasformazioni reversibili, la relazione precedente diviene:
PdV TdS dU = (90)
Questa relazione pu anche essere dedotta direttamente dallequazione (19) ricordando che
per un sistema monocomponente, chiuso e omogeneo dn
i
= 0.
In termini specifici lequazione precedente, che pu essere vista come una combinazione
del primo e del secondo principio, diventa:
Pdv Tds du = (91)
Pur essendo stata sviluppata per sistemi chiusi e trasformazioni reversibili, poich la
relazione precedente coinvolge solo funzioni di stato essa valida in generale anche per
sistemi monocomponente aperti e trasformazioni qualsiasi.
2.5 Gas perfetto
Come noto, sperimentalmente si nota che:
1 lim lim
0 0
= = |
.
|
\
|
Z
RT
Pv
P P
(92)
dove si introdotto il coefficiente di compressibilit, Z = Pv/(RT). Questo comportamento
mostrato nella Figura 11 per alcuni composti.
Questo significa che tutti i fluidi a pressione sufficientemente bassa seguono unequazione
di stato
29
estremamente semplice, cio:
RT Pv = (93)
nota come equazione di stato del gas perfetto. La costante presente in questa equazione,
detta costante dei gas perfetti, vale R = 8,314 [J mol
-1
K
-1
] = 1,987 [cal mol
-1
K
-1
] = 0,082
[atm m
3
kmol
-1
K
-1
].
29
Unequazione di stato una relazione funzionale tra pressione, temperatura e volume molare.
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
39
Figura 11: coefficiente di compressibilit in funzione della pressione per diversi composti.
Fisicamente, un gas perfetto pu essere rappresentato come un fluido cos rarefatto che le
molecole che lo costituiscono non si vedono, cio in cui non esistono interazioni steriche
o energetiche tra le molecole.
Il fatto che un fluido segua lequazione di stato (93) rende particolarmente semplice il
calcolo delle grandezze termodinamiche. Infatti, essendo lenergia interna specifica una
variabile intensiva, essa risulta definita quando sono fissate altre due variabili intensive, per
esempio T e v. In altri termini, si pu dire che u = u(T,v); essendo poi una funzione di stato,
il suo differenziale esatto e pu essere espresso come:
dv
v
u
dT
T
u
du
T v
|
.
|
\
|
+ |
.
|
\
|
=
(94)
Questa relazione ha un chiaro significato fisico: la prima derivata parziale fornisce la
dipendenza dellenergia interna molare dalla temperatura, mentre la seconda derivata
parziale fornisce la dipendenza dellenergia interna molare dal volume molare.
0 5 10 15 20 25 30 35 40 45 50
0.9
0.91
0.92
0.93
0.94
0.95
0.96
0.97
0.98
0.99
1
P [bar]
Z
CH4
Ar
CO
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
40
Mentre la derivata parziale rispetto alla temperatura pari al calore specifico molare a
volume costante, risulta meno immediato valutare il valore della seconda derivata parziale,
quella rispetto al volume molare.
Dalla equazione (91) si pu derivare la seguente relazione:
T T T
v
v
P
v
s
T
v
u
|
.
|
\
|
|
.
|
\
|
= |
.
|
\
|
(95)
Poich si pu dimostrare
30
che:
v T
T
P
v
s
|
.
|
\
|
= |
.
|
\
|
(96)
la relazione precedente diventa:
P
T
P
T
v
u
v T
|
.
|
\
|
= |
.
|
\
|
(97)
Se il fluido segue lequazione di stato del gas perfetto ( ) v R T P
v
/ / = e la relazione
precedente diventa:
0 = = |
.
|
\
|
P
v
RT
v
u
T
(98)
Questo significa che lenergia interna specifica di un gas perfetto non dipende dal volume
specifico ma solo dalla temperatura
31
e la relazione (94) si riduce alla semplice (nel seguito
verranno contrassegnate con un * le grandezze relative a un gas perfetto):
dT c du
* *
V
= (99)
Ovviamente, lenergia interna U
*
= nu
*
dipende sia dalla temperatura sia dal numero di
moli.
Dal fatto che lenergia interna specifica dipende solo dalla temperatura ne consegue che
anche altre grandezze termodinamiche specifiche dipendono solo dalla temperatura, in
particolare entalpia e calore specifico a pressione e volume costante:
) ( ) (
* * * *
T h RT T u Pv u h = + = + = (100)
30
Questo viene fatto in modo molto agevole nel paragrafo 4.1.4 dove vengono discusse le cosiddette
relazioni di Maxwell. Per il momento si accetti questa affermazione senza dimostrazione.
31
Il fatto che dipenda solo dalla temperatura ovviamente implica anche che non dipende nemmeno
dalla pressione.
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
41
) (
*
* *
*
T c
dT
du
T
u
c
V V
v
= =
|
|
.
|
\
|
= (101)
) (
*
* *
*
T c
dT
dh
T
h
c
P P
P
= =
|
|
.
|
\
|
= (102)
dove alle derivate parziali sono state sostituite le derivate totali in quanto le grandezze
dipendono solo dalla temperatura.
Per un gas perfetto la relazione tra le capacit termiche specifiche a pressione e volume
costante particolarmente semplice. Differenziando la definizione di entalpia (100) si
ottiene:
RdT du dh RT u Pv u h + = + = + =
* * * * * *
(103)
Inserendo le relazioni (101) e (102) si ottiene:
R c c RdT dT c dT c
V P V P
+ = + =
* * * *
(104)
La dipendenza dalla pressione dellentropia si conserva anche per un gas perfetto. Dalla
relazione (91) si ricava:
( )
P
dP
R
T
dT
c dP
P
R
T
dT
R c
dP
P
RT
dT
P
R
T
P
T
dT
c dv
T
P
T
dT
c ds
P V
V V
= + =
= |
.
|
\
|
+ = + =
* *
2
* * * *
(105)
dove si utilizzata la relazione derivata dallequazione di stato del gas perfetto:
RdT vdP Pdv = + (106)
Poich si perde la dipendenza dalla pressione, il valore dellentalpia e dellenergia interna
di un gas perfetto si calcola facilmente una volta che definito uno stato di riferimento
dove si pone uguale a zero il valore dellentalpia:
( ) ( )
= + =
T
T
T
T
r
r
P
r
P
dT c dT c T h T h
* * * *
(107)
( ) ( ) ( )
r
T
T
T
T
r r
T
T
r
RT dT c dT c RT T h dT c T u T u
r
V
r
V
r
V
= + = + =
* * * * * *
(108)
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
42
Analogamente, il valore dellentropia si calcola come
32
:
( ) ( )
|
|
.
|
\
|
+ =
r
T
T
P
r r
P
P
R dT
T
c
P T s P T s
r
ln , ,
*
* *
(109)
La relazione (101) implica anche che la variazione di energia interna si calcola sempre
come:
=
2
1
* *
T
T
dT c u
V
(110)
anche per processi non isocori, mentre la relazione (102) consente di calcolare la variazione
di entalpia come:
=
2
1
* *
T
T
P
dT c h
(111)
anche per processi non isobari.
La variazione di entropia si calcola invece come:
|
|
.
|
\
|
=
1
2
*
*
ln
2
1
P
P
R dT
T
c
s
T
T
P
(112)
Per un gas perfetto sono disponibili i calori specifici a pressione costante in funzione della
temperatura, solitamente in forma polinomiale, come riportato a titolo di esempio per alcuni
composti in Tabella 3.
Lesprimere in termini polinomiali la dipendenza dalla temperatura dei calori specifici
consente il calcolo analitico degli integrali presenti nelle relazioni precedenti.
32
Contrariamente ad altre funzioni termodinamiche, possibile calcolare il valore assoluto
dellentropia e non solo le sue variazioni tra due stati. Infatti, da misure sperimentali dei calori
specifici a bassissime temperature si evince che lentropia di diverse forme cristalline della stessa
specie a 0 K assume sempre lo stesso valore, che inferiore al valore assunto da forme non cristalline.
Questo ha portato a postulare che lentropia assoluta di tutte le specie nella forma di cristalli perfetti a
0 K uguale a zero (terza legge della termodinamica). Daltro canto, per applicazioni nellambito
dellingegneria si interessati a variazioni di entropia e quindi possibile scegliere uno stato di
riferimento arbitrario.
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
43
2.5.1 Trasformazioni semplici
Considerando sistemi chiusi e trasformazioni reversibili il primo principio assume la forma
dellequazione (56) che, in termini di grandezze specifiche diventa:
Pdv dq du = (113)
Per trasformazioni isocore di un gas perfetto questa relazione diventa:
= = = =
2
1
* * * *
T
T
V V
dT c u q dT c du dq
(114)
Invece, per trasformazioni isobare si ottiene:
( )
= = = = + = + =
2
1
* * * * * * * *
T
T
P P
dT c h q dT c dh Pv u d Pdv du dq
(115)
Composto T
max
[K] a b x 10
3
c x 10
6
d x 10
9
e x 10
-5
acqua 2000 28,850 12,055 0,000 0,000 1,006
ammoniaca 1800 29,747 25,108 0,000 0,000 -1,546
anidride
carbonica
2000 45,369 8,688 0,000 0,000 -9,619
azoto 2000 27,270 4,930 0,000 0,000 0,333
etano 1000 4,493 182,229 -74,843 10,797 0,000
etilene 1000 4,196 154,565 -81,076 16,813 0,000
idrogeno 3000 27,012 3,509 0,000 0,000 0,690
metano 1000 17,449 60,449 1,117 -7,204 0,000
n-butano 1500 16,087 306,911 -97,796 0,000 0,000
n-ottano 1500 67,900 586,694 -184,637 0,000 0,000
ossido di
carbonio
1500 28,160 1,675 5,372 -2,222 0,000
ossigeno 2000 30,255 4,207 0,000 0,000 -1,887
propano 1000 22,987 176,691 0,000 0,000 0,000
Tabella 3 - Coefficienti per il calcolo dei calori specifici a pressione costante di composti
come gas perfetti.
2 3 2 *
+ + + + = eT dT cT bT a c
P
[J mol
-1
K
-1
]; T in [K]. Per esempio, per
lacqua come gas perfetto a 500 [K] si pu calcolare:
3 , 35 500 10 006 , 1 500 10 055 , 12 850 , 28
2 5 3 *
,
2
= + + =
O H P
c [J mol
-1
K
-1
]. Le correlazioni
sono utilizzabili tra 298 [K] e T
max
.
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
44
Per trasformazioni isoterme, poich per un gas perfetto lenergia interna specifica dipende
solo dalla temperatura:
* * *
0 Pdv dw dq dw dq Pdv dq du = = = + = = (116)
che integrata fornisce:
|
|
.
|
\
|
=
|
|
.
|
\
|
=
|
|
.
|
\
|
= = = =
2
1
1
2
*
*
*
*
*
ln
/
/
ln ln
1
2
2
1
2
1
P
P
RT
P RT
P RT
RT
v
v
RT dv
v
RT
Pdv w q
v
v
v
v
(117)
Per trasformazioni adiabatiche (e quindi, essendo le trasformazioni reversibili, anche
isoentropiche) la variazione di energia interna deve eguagliare il lavoro entrante nel
sistema:
dT c dv
v
RT
Pdv dw du
V
* *
*
*
= = = = (118)
da cui si ottiene:
( )
*
*
*
*
*
1
v
dv
v
dv
c
R
T
dT
V
= = (119)
in cui si introdotto il rapporto tra i calori specifici,
* * * *
/ ) ( /
V V V P
c R c c c + = = . Integrando
la relazione precedente con lapprossimazione di cost si ottiene la seguente relazione:
( )
1
*
2
*
1
1
2
*
1
*
1
2 2
ln 1 ln
|
|
.
|
\
|
=
|
|
.
|
\
|
=
|
|
.
|
\
|
v
v
T
T
v
v
T
T
(120)
Inserendo lequazione di stato del gas perfetto si ottengono le relazioni equivalenti:
1
1
2
1
2
|
|
.
|
\
|
=
P
P
T
T
(121)
*
2 2
*
1 1
v P v P =
(122)
La variazione di energia interna, che pari al lavoro, si ottiene integrando lequazione
(118):
= =
2
1
* *
T
T
V
dT c u w
(123)
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
45
Se il calore specifico pu essere considerato costante (cosa ragionevole per piccole
variazioni di temperatura) la relazione precedente diviene:
( ) ( )
1 1
*
1 1
*
2 2
1 2 1 2
* *
= = =
v P v P
T T
R
T T c u w
V
(124)
Inserendo la relazione (122) e utilizzando lequazione di stato del gas perfetto si pu
ottenere, attraverso alcuni passaggi algebrici, la nuova relazione:
|
|
|
.
|
\
|
|
|
.
|
\
|
= =
1
1
1
1
2 1 *
P
P RT
u w (125)
Limportanza delle funzioni di stato nella termodinamica pu essere compresa dalla
seguente generalizzazione. Tutte le relazioni discusse in questa sezione sono state
sviluppate per sistemi chiusi, processi reversibili e gas perfetto. Poich il valore delle
funzioni di stato dipende solo dallo stato considerato e non dalle trasformazioni seguite per
raggiungere tale stato, tali relazioni valgono anche per sistemi aperti e trasformazioni
irreversibili di un gas perfetto. Viceversa, le relazioni sviluppate per il calore e il lavoro
scambiato (che non sono funzioni di stato) non possono essere generalizzate a sistemi aperti
e trasformazioni irreversibili.
Questa generalizzazione una procedura molto comune in termodinamica. Poich la
variazione del valore di una funzione di stato tra due stati non dipende dalla trasformazione
considerata, tale variazione viene sempre calcolata utilizzando la trasformazione pi
comoda per i calcoli.
2.5.2 Applicazione
Un gas perfetto viene compresso adiabaticamente e reversibilmente in un cilindro in cui
inizialmente la temperatura pari a 300 K e la pressione a 0,1 MPa fino a ridurne il volume
a 1/15 del valore iniziale. Sapendo che per il gas considerato 2 , 1 = si calcoli la
temperatura e la pressione finale del gas, oltre al lavoro specifico necessario per la
compressione.
Dalla relazione (122) si pu ricavare il valore finale della pressione:
[MPa] 58 , 2
1
15
1 , 0
2 , 1
2
1
1
*
2
*
1
1 2
= |
.
|
\
|
=
|
|
.
|
\
|
=
|
|
.
|
\
|
=
V
V
P
v
v
P P (126)
Si noti che poich il sistema chiuso il numero di moli non varia, e quindi il rapporto tra i
volumi molari uguale al rapporto tra i volumi del sistema.
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
46
La temperatura finale pu essere calcolata dalla equazione di stato del gas perfetto applicata
allo stato iniziale e finale:
[K] 516
15 1 , 0
1 58 , 2
300
1 1
2 2
1
*
1 1
*
2 2
1 2
2
*
2 2
1
*
1 1
=
= = = = =
V P
V P
T
v P
v P
T T
T
v P
R
T
v P
(127)
Il lavoro scambiato viene calcolato utilizzando la relazione (125):
] mol [kJ 97 , 8 ] mol [J 8966 1
1 , 0
58 , 2
1 2 , 1
300 314 , 8
1 1
2 , 1
1 2 , 1
= =
|
|
|
.
|
\
|
|
|
.
|
\
|
= w (128)
Essendo il valore positivo si tratta di lavoro entrante nel sistema, cio che deve essere
fornito per comprimere il gas.
Gli stessi risultati possono essere ovviamente ottenuti utilizzando i bilanci di materia e di
energia applicati a un sistema definito da una superficie di controllo variabile nel tempo e
tale da contenere il gas. Infatti, le relazioni utilizzate, come discusso in precedenza,
derivano da tali bilanci. Si lascia allo studente lo sviluppo dellapplicazione con questo
approccio discusso nellApplicazione seguente.
2.6 Applicazione
Una bombola da 60 [l] viene riempita di aria fino alla pressione di 40 [bar] collegandola
con una linea di aria compressa a 50 [bar] e 20 [C]. La pressurizzazione della bombola
cos rapida che la si pu assumere adiabatica. Se la bombola contiene inizialmente aria a 1
[bar] e 20 [C], quale sar la temperatura alla fine della carica? Dopo un certo periodo di
tempo il gas nella bombola scambia calore con lambiente e si porta alla temperatura di 20
[C]: a che pressione si porta? Si consideri laria un gas perfetto con 21
*
=
V
c [J mol
-1
K
-1
].
Per risolvere un problema nellambito dellingegneria necessario che sia ben posto, cio
che il numero di incognite del problema sia pari al numero di equazioni indipendenti che le
correlano. Una volta scritto un numero di equazioni pari al numero di incognite, la
risoluzione un semplice problema algoritmico
33
.
33
La ricerca dellalgoritmo pi opportuno per la risoluzione di un dato problema pu essere anche
non banale, soprattutto se il sistema di equazioni non ammette una soluzione analitica. Si tratta per
di un problema diverso rispetto alla corretta scrittura delle equazioni da risolvere. I due aspetti della
risoluzione di un problema (la definizione di un numero di equazioni indipendenti pari al numero
delle incognite e la loro successiva risoluzione) non devono essere confusi: nessuna manipolazione di
un numero di equazioni insufficiente (cio inferiore al numero delle incognite) potr mai portare alla
soluzione del problema.
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
47
Considerando una superficie di controllo che racchiude il gas presente nella bombola come
schematizzato in Figura 12 si possono scrivere ed integrare tra linizio (stato 1) e la fine
(stato 2) della carica i bilanci di materia e di energia:
= =
F
t
IN IN
dt n n n n
dt
dn
0
1 2
& & (129)
= =
= =
= =
F F
t
IN IN
t
IN IN
IN IN IN
dt n T h dt T h n
T u n T u n U U
T h n H
dt
dU
0
*
0
*
1
*
1 2
*
2 1 2
*
) ( ) (
) ( ) (
) (
& &
&
&
(130)
Figura 12: superficie di controllo per la carica della bombola.
Le incognite che compaiono in queste due equazioni sono
IN
n& , n
1
, n
2
e T
2
. Si noti che la
temperatura iniziale e quella dellaria in ingresso sono note, e che lentalpia e lenergia
interna specifica possono essere calcolate con le relazioni discusse in precedenza in
funzione della sola temperatura. Si hanno quindi 2 equazioni in 4 incognite: necessario
superficie di
controllo
linea di
carico
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
48
formulare altre 2 equazioni indipendenti, quali le equazioni di stato del gas perfetto nelle
condizioni iniziali e finali:
2 2 2
1 1 1
RT n V P
RT n V P
=
=
(131)
Essendo noto il volume, si hanno ora 4 equazioni in 4 incognite ed il problema risolubile.
Un possibile algoritmo di soluzione descritto nel seguito.
Sostituendo la relazione (129) nella (130) si ottiene:
( )
1 2
*
1
*
1 2
*
2
) ( ) ( ) ( n n T h T u n T u n
IN
= (132)
introducendo le relazioni per il calcolo di entalpia ed energia interna (T
r
la temperatura di
riferimento arbitraria) si ottiene:
( ) ( ) ( ) ( ) ( )( )
1 2
*
1
*
1 2
*
2
n n T T c RT T T c n RT T T c n
r IN P r r V r r V
= (133)
Inserendo la relazione (104) con alcuni passaggi algebrici si ottiene:
( ) ( ) ( )
1 2
*
1
*
1 2
*
2 1 2
* *
1
*
2
n n T c T c n T c n n n c c n c n T
IN P V V P P P r
= + + + (134)
Si nota che, correttamente, tutti i termini contenenti lo stato di riferimento si elidono per
dare una relazione che non dipende dallo stato di riferimento arbitrariamente scelto:
( )
1 2
*
1
*
1 2
*
2
n n T c T c n T c n
IN P V V
= (135)
Ne consegue che solitamente conveniente, per semplificare la scrittura dellequazione,
scegliere come temperatura di riferimento quella di uno degli stati coinvolti nellequazione.
Per esempio, scegliendo come temperatura di riferimento quella della corrente entrante, T
IN
,
lentalpia della corrente entrante nulla e lequazione (132) diventa:
( ) ( ) ( ) ( ) 0
1
*
1 2
*
2
=
IN IN V IN IN V
RT T T c n RT T T c n (136)
che, ricordando la relazione (104), risulta identica alla (135).
Inserendo le due equazioni (131) non ancora utilizzate nella (135) si ottiene:
|
|
.
|
\
|
=
1
1
2
2 *
1
*
1
1
2
*
2
2
RT
V P
RT
V P
T c T c
RT
V P
T c
RT
V P
IN P V V
(137)
che semplificata e risolta rispetto allunica incognita presente, T
2
, fornisce:
[K] 405
293
1 40
314 , 8 21
21
293
1
40
1 2
*
*
1
1
2
2
=
|
.
|
\
|
+
+
=
|
|
.
|
\
|
+
=
IN P
V
T
P P
c
c
T
P
P
T
(138)
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
49
Al termine del raffreddamento (stato 3) lequazione di stato del gas perfetto :
3 3
nRT V P = (139)
che rappresenta una equazione nelle due incognite P
3
e n. La seconda equazione necessaria
per risolvere il problema sempre lequazione di stato del gas perfetto scritta per lo stato 2,
cio al termine della carica. In questo stato il numero di moli presenti lo stesso di quello
presente nello stato 3 e quindi:
2 2
nRT V P = (140)
Il sistema di 2 equazioni in 2 incognite pu ora essere risolto. Un possibile algoritmo di
soluzione prevede di dividere membro a membro le due equazioni, semplificarle ed
esplicitare lunica incognita rimasta, P
3
:
[bar] 9 , 28
405
293
40
2
3
2 3
= = =
T
T
P P
(141)
2.7 Esercizi
Dellacqua a 200 [F] viene pompata da un serbatoio alla portata di 50 [gal min
-1
]. Il motore
della pompa fornisce una potenza di 2 [hp]. Dopo la pompa, lacqua passa attraverso uno
scambiatore di calore assorbendo 40000 [Btu min
-1
] e viene infine scaricata in un secondo
serbatoio posto 50 [ft] pi in alto del primo. Si calcoli la temperatura a cui viene scaricata
lacqua trascurando la dipendenza dellentalpia dalla pressione e considerando
P
c costante
e pari a 1 [cal g
-1
K
-1
].
Risultato: 147 [C].
Un sistema chiuso passa dallo stato 1 allo stato 2 attraverso una trasformazione A ricevendo
100 [J] di calore e fornendo 40 [J] di lavoro. Lo stesso sistema passa dallo stato 1 allo stato
2 attraverso una trasformazione B fornendo 40 [J] di lavoro. Quale il valore del calore
scambiato nella seconda trasformazione? Il calore entra o esce dal sistema?
Risultato: 80 [J]; il calore entra nel sistema.
Un serbatoio di 0,3 [m
3
] contiene aria a 10 [bar] e 330 [K] e viene svuotato fino a pressione
atmosferica. Si calcoli la temperatura e il numero di moli al termine dello scarico se:
a) si mantiene la temperatura costante;
b) si impedisce ogni scambio termico con lambiente.
Si consideri laria un gas perfetto con
*
P
c = 29 [J mol
-1
K
-1
],
Risultato: a) 330 [K]; 11,1 [mol]. b) 171 [K]; 21,4 [mol].
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
50
Un compressore aspira aria in quiete a 100 [kPa] e 298 [K] e la scarica a 1000 [kPa] e 340
[K]. Assumendo la velocit allo scarico pari a 60 [m s
-1
] e che il compressore lavori
adiabaticamente, calcolare la potenza richiesta per comprimere 100 [kg h
-1
] di aria. Si
consideri laria un gas perfetto con un calore specifico a pressione costante pari a
2 -3 *
4 , 13302 10 4,780 27,894
+ = T T c
P
[J mol
-1
K
-1
].
Risultato: 1,3 [kW].
Un serbatoio di 0,5 [m
3
] contiene azoto a 17,5 [bar] e 22 [C] che viene scaricato attraverso
un opportuno motore collegato direttamente al serbatoio in modo tale che alluscita dal
motore lazoto sia sempre a 1 [bar] e 22 [C]. Il processo di scarico continua finch la
pressione nel serbatoio raggiunge il valore di 1 [bar]. Sia il serbatoio sia il motore sono
adiabatici e lazoto in queste condizioni pu essere considerato un gas perfetto con
*
P
c = 30
[J mol
-1
K
-1
]. Calcolare:
a) la temperatura finale dellaria nel serbatoio;
b) il lavoro fornito dal motore;
Risultato: a) 133,5 [K]; b) 606,5 [kJ].
Una turbina adiabatica lavora con un gas perfetto a temperatura e pressione di ingresso e
pressione di scarico costanti. Dimostrare che la minima temperatura allo scarico si ha
quando la trasformazione reversibile.
Risultato: dal bilancio di entropia,
|
|
.
|
\
|
+ =
|
|
.
|
\
|
IN
OUT
P P
S
IN
OUT
P
P
c
R
c
R
T
T
ln ln .
Una bombola da 10 [l] contenete azoto come gas perfetto con
*
P
c = 30 [J mol
-1
K
-1
] a 200
[bar] e 250 [K] viene connessa a una seconda bombola di 5 [l] inizialmente vuota finch le
pressioni nelle due bombole diventano uguali. Calcolare la temperatura e pressione finale
nelle due bombole assumendo lintero processo adiabatico.
Risultato: 223,4 [K]; 328,0 [K]; 133,3 [bar].
Si vuole alzare la temperatura di 1 [kmol] di anidride carbonica da 298 [K] a 600 [K]
mantenendo la pressione costante. Calcolare la quantit di calore da fornire. Si consideri il
gas perfetto.
Risultato: 13,255 [MJ].
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
51
Un serbatoio rigido ed isolato da 2 [m
3
] contiene un gas perfetto (
*
P
c = 2,5 R) separato in
due comparti uguali: il primo si trova a 400 [K] e 3 [MPa], il secondo a 600 [K] e 1 [MPa].
Calcolare la variazione di entropia conseguente alla rottura del setto di separazione dei due
compartimenti.
Risultato: 2,19 [kJ K
-1
].
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
53
3 Comportamenti volumetrici dei fluidi
puri
Sperimentalmente si verifica che per un sistema monocomponente e monofase sufficiente
fissare due variabili intensive per definirne lo stato intensivo. Considerando le tre variabili
intensive misurabili: pressione, temperatura e volume specifico, deve quindi esistere una
relazione tra queste tre variabili che consenta di calcolarne una quando le altre due sono
fissate. Una tale relazione, che pu essere rappresentata in grafici, tabelle o tramite
equazioni matematiche, si chiama equazione di stato
34
e rappresenta largomento principale
di questo capitolo.
Al termine dello studio di questo capitolo lo studente dovrebbe conoscere:
landamento qualitativo dei diagrammi P - v - T dei fluidi puri;
il concetto di transizione di fase;
i calori latenti associati alle transizioni di fase;
il punto critico e il punto triplo;
la tensione di vapore;
il titolo di una miscela bifase;
come calcolare le propriet di una miscela bifase;
il concetto di vapore e liquido saturo;
landamento dellisoterma critica;
la varianza di un sistema monocomponente;
luso delle tabelle e dei diagrammi di stato;
lequazione di stato di van der Waals;
le equazioni di stato cubiche;
landamento di unisoterma calcolata con unequazione di stato cubica nel piano P - v
al variare della temperatura;
quali radici di unequazione di stato cubica hanno un significato fisico;
come calcolare il coefficiente di compressibilit di gas e liquidi con unequazione di
stato cubica;
come calcolare i parametri di unequazione di stato cubica dalla temperatura e
pressione ridotta;
il fattore acentrico di Pitzer;
le variabili ridotte;
lequazione degli stati corrispondenti a due e tre parametri;
lequazione di stato del viriale;
34
Il termine equazione di stato viene spesso indicato con lacronimo anglosassone EoS, Equation of
State.
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
54
3.1 Comportamenti qualitativi
Se si considera una sostanza solida ad un certo valore di temperatura e pressione a cui viene
fornito calore a pressione costante, landamento qualitativo della temperatura e del volume
specifico quello illustrato in Figura 13.
Figura 13: andamento qualitativo della temperatura e del volume specifico durante il
riscaldamento isobaro di una sostanza pura.
La sostanza solida alla temperatura e pressione di partenza rappresentata dal punto 1.
Fornendo calore, la temperatura e il volume specifico del corpo aumentano fino a
raggiungere il punto 2, dove il composto inizia a fondere: si ha una transizione di fase
1
2
6
5
4
3
T
[
K
]
v [m
3
mol
-1
]
vapore
liquido
solido
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
55
caratterizzata dalla simultanea presenza della fase solida e liquida in equilibrio
35
. La fase
solida ha un volume specifico pari a quello del punto 2, mentre la fase liquida che si forma
ha un volume specifico caratterizzato dal punto 3. Poich le due fasi sono in equilibrio
devono avere la stessa temperatura, e quindi la linea che collega il punto 2 al punto 3
orizzontale. In altri termini, le transizioni di fase dei composti puri sono isotermobariche,
cio avvengono a temperatura e pressione costante. Questo implica anche che, a pressione
assegnata, la temperatura di fusione di un composto puro (punto 2) e quella di
solidificazione (punto 3) coincidono: a pressione assegnata, esiste una e una sola
temperatura a cui possono coesistere la fase solida e quella liquida, chiamata temperatura
di fusione o di solidificazione. Ovviamente, a temperatura assegnata esiste una e una sola
pressione a cui possono coesistere la fase solida e quella liquida, chiamata pressione di
fusione o di solidificazione.
Continuando a fornire calore la quantit di sostanza fusa man mano aumenta, e quindi
anche il volume specifico della miscela bifase (cio il volume occupato da una mole di
composto in parte solido e in parte liquido) aumenta poich il volume specifico del liquido
solitamente superiore a quello del solido. Il volume specifico della miscela bifase ha un
valore compreso tra quello del solido e quello del liquido in equilibrio (punti 2 e 3) e quindi
rappresentato da un punto sul segmento 2-3. In altri termini, la regione compresa tra i
punti 2 e 3 rappresenta la regione bifase solido - liquido. Il valore del volume occupato da
una mole di composto in parte liquido e in parte solido pu essere calcolato dai valori del
volume specifico del solido e del liquido in equilibrio con la relazione:
( )
S S L L S L
v n v n v n n + = + (142)
da cui si ricava:
( ) ( )
( )
S L S
S L
S
L
S L
L
v x xv v
n n
n
v
n n
n
v + =
+
+
+
= 1 (143)
dove si inserito il titolo in liquido della miscela, definito come ( )
S L L
n n n x + = . Questa
relazione di carattere generale e consente il calcolo di una generica grandezza specifica
della miscela, m, come:
( )
S L
m x xm m + = 1 (144)
La quantit di calore che deve essere fornita per fondere completamente una mole di
composto prende il nome di calore latente di fusione. Poich tale calore viene fornito a
pressione costante, prende anche il nome di entalpia di fusione. Il calore latente (o
35
Si parla di due fasi distinte in quanto le propriet delle due fasi, per esempio il volume specifico,
sono differenti.
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
56
entalpia) di solidificazione coincide col calore latente di fusione a parte il segno, poich in
questo caso bisogna sottrarre calore al sistema anzich fornirlo.
Quando tutta la sostanza fusa (punto 3) il calore fornito provoca un aumento della
temperatura e del volume specifico fino a raggiungere il punto 4, dove il composto inizia a
bollire. A questo punto si ha la simultanea presenza della fase vapore e liquida in equilibrio.
La fase vapore ha un volume specifico pari a quello del punto 5, mentre la fase liquida ha
un volume specifico caratterizzato dal punto 4. Poich le due fasi sono in equilibrio devono
avere la stessa temperatura, e quindi la linea che collega il punto 4 al punto 5 orizzontale
(cio la transizioni di fase isotermobarica). Questo implica che, a pressione assegnata, la
temperatura di ebollizione di un composto puro (punto 4) e quella di solidificazione (punto
5) coincidono: a pressione assegnata, esiste una e una sola temperatura a cui possono
coesistere la fase vapore e quella liquida, chiamata temperatura di ebollizione o di
condensazione. Ovviamente, a temperatura assegnata esiste una e una sola pressione a cui
possono coesistere la fase solida e quella liquida, chiamata pressione (o tensione) di
vapore.
Continuando a fornire calore la quantit di vapore man mano aumenta, e quindi anche il
volume specifico della miscela bifase (cio il volume occupato da una mole di composto in
parte liquido e in parte vapore) aumenta poich il volume specifico del vapore superiore a
quello del liquido. Il volume specifico della miscela bifase ha un valore compreso tra quello
del liquido e quello del vapore in equilibrio (punti 4 e 5) e quindi rappresentato da un
punto sul segmento 4-5. In altri termini, la regione compresa tra i punti 4 e 5 rappresenta la
regione bifase liquido - vapore. Il valore del volume occupato da una mole di composto in
parte liquido e in parte vapore pu essere calcolato da una relazione analoga alla (4):
( )
L L V V V L
v n v n v n n + = + (145)
da cui si ricava:
( ) ( )
( )
L V L
V L
L
V
V L
V
v x xv v
n n
n
v
n n
n
v + =
+
+
+
= 1 (146)
dove si inserito il titolo in vapore della miscela, definito come ( )
V L V
n n n x + = . Anche
questa relazione di carattere generale e consente il calcolo di una generica grandezza
specifica della miscela, m, come:
( )
L V
m x xm m + = 1 (147)
La quantit di calore che deve essere fornita per evaporare completamente una mole di
composto prende il nome di calore latente (o entalpia) di evaporazione che coincide col
calore latente di condensazione a parte il segno.
Fornendo ulteriore calore il vapore si riscalda aumentando la sua temperatura e il suo
volume specifico fino al punto 6.
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
57
Variando il valore della pressione il comportamento qualitativo rimane immutato, anche se
ovviamente la curva 1-2-3-4-5-6 risulta traslata verso lalto (aumento della pressione) o
verso il basso (riduzione della pressione).
Gli stessi comportamenti vengono solitamente rappresentati su un diagramma P - v, come
illustrato in Figura 14 solo per la porzione relativa alle fasi liquida e vapore.
Figura 14: andamento qualitativo della pressione e del volume specifico durante la
compressione isoterma di una sostanza pura.
Si consideri una fase liquida contenuta in un cilindro mantenuto a temperatura costante e
chiuso da un pistone mobile (punto 1 nella figura). Alzando il pistone il volume aumenta e
la pressione diminuisce. Essendo il sistema chiuso in numero di moli rimane costante a
laumento di volume implica anche un aumento del volume specifico. Si noti che al
diminuire della pressione il volume specifico aumenta di poco in quanto i liquidi sono
praticamente incomprimibili
36
. Questo comportamento procede fino al punto 2, dove il
36
Questa una caratteristica generale dei liquidi lontani dal punto critico: le propriet dei liquidi sono
poco influenzate dalla pressione e possono quindi essere considerate con buona approssimazione
costanti al variare della pressione stessa. Lo stesso vale per le propriet dei solidi.
1
2
4
3
P
[
b
a
r
]
v [m
3
mol
-1
]
vapore
liquido
curva del
vapore saturo
curva del
liquido saturo
punto critico
T
1
T
2
> T
1
T
C
> T
2
T
3
> T
C
v
V
v
L
P(T
1
)
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
58
liquido inizia a bollire. A questo punto si ha la simultanea presenza della fase vapore e
liquida in equilibrio. La fase vapore ha un volume specifico pari a quello del punto 3,
mentre la fase liquida ha un volume specifico caratterizzato dal punto 2. Poich le due fasi
sono in equilibrio devono avere la stessa temperatura, e quindi la linea che collega il punto
2 al punto 3 orizzontale. Il valore di pressione a cui esistono le due fasi in equilibrio la
pressione (o tensione) di vapore alla temperatura considerata, indicata come P(T
1
) nella
figura.
Continuando ad aumentare il volume la quantit di vapore man mano aumenta, ma, essendo
la temperatura costante, la pressione non varia finch sono presenti entrambe le fasi.
Aumentando il volume aumenta per il volume specifico della miscela bifase, che ha un
valore compreso tra quello del liquido e quello del vapore in equilibrio (punti 2 e 3) e
quindi rappresentato da un punto sul segmento 2-3. In altri termini, analogamente al
diagramma T - v discusso in precedenza, la regione compresa tra i punti 2 e 3 rappresenta la
regione bifase liquido - vapore e i punti 2 e 3 sono caratteristici del vapore saturo e del
liquido saturo, cio del vapore e del liquido in equilibro.
Quando tutto il liquido evaporato si ha solo vapore (punto 3): continuando ad alzare il
pistone il volume specifico aumenta e la pressione riprende a diminuire (punto 4). Si noti
che al diminuire della pressione il volume specifico aumenta sensibilmente in quanto i
vapori sono molto comprimibili.
Ripetendo lesperimento a una temperatura superiore (indicata con T
2
nella figura) il
comportamento qualitativo analogo, ma la curva 1-2-3-4 risulta traslata verso lalto. Si
nota per che in corrispondenza della transizione di fase la variazione del volume specifico
tra il liquido saturo e il vapore saturo risulta meno marcata. Questo comportamento
prosegue aumentando ulteriormente la temperatura finch la differenza tra il volume
specifico delle due fasi diventa infinitamente piccolo fino a scomparire in corrispondenza di
un particolare valore della temperatura, detta temperatura critica. A questa temperatura non
si ha pi la transizione di fase. Poich il liquido e il vapore in equilibrio sono collegati da
un segmento orizzontale che diventa via via pi piccolo avvicinandosi alla temperatura
critica, intuitivo comprendere che lisoterma critica deve presentare un flesso a tangente
orizzontale in corrispondenza del punto critico:
0
,
2
2
,
=
|
|
.
|
\
|
= |
.
|
\
|
c c
c c P T
P T
v
P
v
P
(148)
La pressione relativa al punto critico detta pressione critica. I valori della temperatura e
pressione critica sono caratteristici di ciascun composto e sono facilmente reperibili nei
manuali tecnici
37
. I valori per alcune sostanze sono riportati in Tabella 4.
37
Si veda per esempio il manuale dellingegnere chimico: R.H. Perry, D.W. Green, Perrys Chemical
Engineers Handbook, McGraw-Hill, NY (1999).
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
59
Per temperature superiori alla temperatura critica non si presenta pi la transizione di fase.
Ripetendo lesperimento precedente a una temperatura superiore a quella critica (T
3
nella
figura), il volume specifico aumenta e la pressione diminuisce in modo monotono.
Composto
Temperatura
critica [K]
Pressione
critica [bar]
Coefficiente di
compressibilit
critico [-]
Fattore
acentrico [-]
acqua 647,30 221,2 0,230 0,344
ammoniaca 405,50 112,77 0,243 0,250
anidride
carbonica
304,20 73,87 0,275 0,239
azoto 126,20 33,94 0,291 0,039
benzene 562,10 49,24 0,274 0,212
cloro 417,00 77,11 0,276 0,090
etano 305,43 48,84 0,285 0,099
etilene 283,10 51,17 0,270 0,089
metano 190,70 46,41 0,290 0,011
monossido di
carbonio
133,00 34,96 0,294 0,066
n-butano 425,20 37,97 0,274 0,199
n-ottano 569,40 24,97 0,256 0,398
ossigeno 154,40 50,36 0,290 0,025
Tabella 4: valori della temperatura, pressione e coefficiente di compressibilit nel punto
critico, e valori del fattore acentrico.
Il luogo dei punti caratteristici del vapore saturo viene chiamato curva del vapore saturo,
mentre quello dei punti caratteristici del liquido saturo viene detto curva del liquido saturo.
Entrambe le curve nascono dal punto critico, come mostrato in figura, e racchiudono la
regione bifase. La loro unione viene solitamente chiamata campana di Andrews
38
.
Infine, le stesse informazioni possono essere riportate su un diagramma P - T, come
illustrato in Figura 15.
Le tre curve rappresentano i confini di esistenza delle tre fasi (solida, liquida e vapore):
attraversando ciascuna di queste curve si ha una transizione di fase. Considerando per
esempio la curva di evaporazione, a temperatura T
1
per pressioni inferiori a P(T
1
), che
rappresenta la tensione di vapore a temperatura T
1
per il composto in esame, esiste solo la
fase vapore. A valori superiori a P(T
1
) esiste solo la fase liquida. Solo e soltanto quando la
pressione pari a P(T
1
) possono coesistere la fase liquida e vapore in equilibrio.
Considerazioni analoghe possono essere fatte per la curva di sublimazione (equilibrio
38
Thomas Andrews, 1813 - 85, fisico e chimico irlandese.
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
60
vapore - solido) e di fusione (equilibrio liquido - solido). Sopra la temperatura critica non si
ha pi transizione di fase e la curva di equilibrio si interrompe.
Figura 15: confini delle fasi per una sostanza pura.
Nella Figura 15 anche indicato il punto triplo, cio lunico valore di temperatura e
pressione a cui possono coesistere le tre fasi in equilibrio.
Si ripetutamente ricordato che per caratterizzare completamente lo stato intensivo di un
sistema monocomponente e monofase sufficiente assegnare due variabili intensive, in
quanto tutte le altre risultano fissate. Poich ora noto che due fasi possono coesistere in
equilibrio a una data temperatura solo per un dato valore di pressione, evidente che
sufficiente assegnare una sola variabile intensiva per caratterizzare lo stato intensivo di un
P
[
b
a
r
]
T [K]
vapore
liquido
curva di
evaporazione
curva di
sublimazione
l li id
punto critico
curva di
fusione
T
C
P
C
solido
punto
triplo
T
1
P(T
1
)
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
61
sistema monocomponente e bifase. Per esempio, assegnata la temperatura T
1
in Figura 15,
univocamente definita la pressione P(T
1
) a cui possono coesistere la fase liquida e vapore,
e dalla Figura 14 i valori dei volumi specifici delle due fasi in equilibrio (v
L
e v
V
). Se poi si
vuole caratterizzare lo stato intensivo di un sistema monocomponente trifase, non possiamo
assegnare arbitrariamente il valore a nessuna variabile intensiva in quanto vi un unico
valore di temperatura e pressione dove coesistono le tre fasi in equilibrio. Quanto detto
viene riassunto nella definizione di varianza (o gradi di libert), che altro non che il
numero di variabili intensive che devono essere fissate per definire in modo univoco lo
stato intensivo di un sistema
39
. Per un sistema monocomponente la varianza pu quindi
essere calcolato come:
F V = 3 (149)
dove F il numero di fasi presenti nel sistema.
3.1.1 Applicazione
Un recipiente chiuso e rigido contenente acqua sotto forma di una miscela di vapore e
liquido saturo a 100 [C] viene riscaldato fino a raggiungere il punto critico. Noto il volume
specifico dellacqua al punto critico (3,170 10
-3
[m
3
kg
-1
]), del liquido saturo a 100 [C]
(1,044 10
-3
[m
3
kg
-1
]) e del vapore saturo a 100 [C] (1,673 [m
3
kg
-1
]), calcolare il rapporto
tra il volume di liquido e quello di vapore inizialmente presente nel recipiente.
Poich il recipiente chiuso e rigido la massa presente nel sistema e il volume non cambia.
Di conseguenza, anche il volume massico rimane invariato nel corso della trasformazione:
( ) ( ) ( ) x x v x v x v P T v
L V
IN C C
+ = + = = =
1 10 044 , 1 673 , 1 1 10 170 , 3 ,
3 3
(150)
da cui si ricava il titolo in vapore, x = 1,27 10
-3
.
Il rapporto tra il volume di vapore e liquido si calcola quindi come:
( )
( )
] m [m 04 , 2
10 044 , 1 10 27 , 1 1
673 , 1 10 27 , 1
] kg [m ] kg [kg 1
] kg [m ] kg [kg
3
L
3
V
3 3
3
1 -
L
3
L
1
tot L
-1
V
3
V
1
tot V
=
=
=
=
L
V
L
V
v x
v x
V
V
(151)
39
Questo significa che tutte le altre variabili intensive possono assumere solo un ben definito valore,
che deve essere calcolabile dai valori delle variabili intensive fissate.
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
62
3.2 Diagrammi di stato e tabelle
Il modo pi semplice ed immediato per rappresentare la relazione f(T,P,v) = 0, cio
lequazione di stato di una sostanza, quello di riportare su di un grafico o in una tabella il
valore del volume specifico in funzione della pressione e della temperatura.
Per esempio, in Appendice B sono riportate le tabelle per lacqua. Per una coppia di valori
di temperatura e pressione, la tabella riporta il valore del volume specifico del vapore saturo
e del liquido saturo. Poich la pressione influenza poco il volume specifico dei liquidi
lontano dal punto critico, il valore del volume specifico del liquido saturo ad una certa
temperatura anche una ragionevole approssimazione del valore del volume specifico del
liquido alla stessa temperatura e a una pressione diversa dalla tensione di vapore a quella
temperatura.
Figura 16: diagramma di stato del n-ottano. La figura riporta quattro curve isoterme e la
campana di Andrews. Per la temperatura di 495 [K] vengono anche indicati i valori della
tensione di vapore e del volume specifico del vapore saturo e del liquido saturo.
10
-1
10
0
10
1
0
5
10
15
20
25
30
v [m
3
kmol
-1
]
P
[
b
a
r
]
495 [K]
540 [K]
580 [K] 620 [K]
P (495)
v
V
sat
v
L
sat
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
63
La Figura 16 riporta invece il diagramma di stato del n-ottano (per semplificare il grafico
sono riportate solo quattro curve isoterme). Noti due valori tra temperatura, pressione e
volume specifico immediato ricavare il terzo.
Il principale problema nelluso di grafici e tabelle che i dati sono riportati in forma
discreta. Quando il valore di interesse non contemplato nei dati della tabella o del grafico
nasce quindi il problema di interpolare tra i due valori adiacenti. Per i diagrammi
linterpolazione non pu che essere di tipo grafico, mentre per le tabelle una semplice
interpolazione lineare solitamente risulta sufficientemente accurata
40
.
3.3 Relazioni matematiche
Il modo pi comodo per rappresentare lequazione di stato di un composto quello di
utilizzare una relazione matematica. A questo fine, sono state proposte molte relazioni
matematiche, ciascuna delle quali presenta alcuni pregi e alcuni difetti. Lutilizzo di una o
laltra relazione dipende dalle condizioni operative considerate e dagli obiettivi del calcolo.
La pi semplice equazione di stato quella del gas perfetto, che gi stata discussa nel
paragrafo 2.5 e rappresenta ragionevolmente bene il comportamento volumetrico dei gas a
bassa pressione. Nel seguito si riportano alcune delle equazioni di stato pi utilizzate.
3.3.1 Equazione di van der Waals e equazioni cubiche
Nel 1873 van der Waals
41
propose unequazione di stato sviluppata partendo da quella dei
gas perfetti tenendo conto del volume che non disponibile per il movimento delle
molecole
42
(in quanto occupato dalle molecole stesse) e delle forze di attrazione tra le
molecole
43
.
Mentre il volume occupato dalle molecole riduce il volume realmente disponibile per il
movimento delle molecole, la presenza di forze di attrazione riduce la pressione esercitata
dal fluido rispetto al caso del gas perfetto. Infatti, una molecola posta in prossimit della
parete del recipiente che contiene il fluido verr attratta dalle altre molecole e quindi
risentir di una forza netta diretta verso linterno del recipiente. Come conseguenza, la
40
Dati due valori tabulati (x
1
< x
2
) a cui corrispondono due valori (y
1
e y
2
), il valore di y
corrispondente a un valore di x compreso tra x
1
e x
2
si calcola come (si veda lAppendice A)
( )
1
1 2
1 2
1
x x
x x
y y
y y
+ =
41
Johannes Diderik van der Waals, 1837 - 1923, fisico olandese, premio Nobel nel 1910.
42
Tale volume escluso, introdotto precedentemente da Clausius, solitamente chiamato covolume.
43
Si tratta di forze di attrazione deboli che agiscono su lunghe distanze, chiamate appunto forze di
van der Waals.
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
64
pressione esercitata da tale molecola sulle pareti del recipiente risulta ridotta rispetto al caso
in cui non vi siano forze di attrazione tra le molecole (come per il gas perfetto).
La forma dellequazione di stato che ne risulta la seguente:
( ) RT b v
v
a
P = |
.
|
\
|
+
2
(152)
dove a e b sono due costanti caratteristiche di un dato composto da determinare per
confronto con misure P - v - T . Lequazione precedente pu essere posta nella forma pi
nota:
2
v
a
b v
RT
P
= (153)
oppure nella seguente:
( ) 0
2 3
= + + ab av v RT Pb Pv (154)
che mette in evidenza come si tratti di unequazione cubica nel volume. Assegnate quindi la
temperatura e la pressione, la soluzione di questa equazione fornisce tre valori del volume,
di cui solo quelli reali positivi possono avere senso fisico.
Come illustrato in Figura 17, quando la temperatura superiore alla temperatura critica
lequazione precedente fornisce una sola radice reale positiva per qualsiasi valore della
pressione.
Viceversa, quando la temperatura inferiore a quella critica, a seconda della pressione
considerata si pu trovare una sola radice positiva (come nel caso a 450 [K] e 15 [bar]
riportato in figura), oppure tre radici reali distinte (come nel caso a 450 [K] e 5 o 10 [bar]
riportato in figura). In questo caso bisogna decidere quale delle radici ha senso fisico
44
. Nel
caso in cui la pressione considerata sia superiore alla tensione di vapore alla temperatura
data esiste solo la fase liquida e lunica radice a cui si pu associare un significato fisico
(quello di volume specifico del liquido) la minore, essendo lunica esterna alla campana
di Andrews (si veda la figura per il caso a 450 [K] e 10 [bar] > 9 ) [K] 450 ( P [bar]). Nel
caso in cui la pressione considerata sia invece inferiore alla tensione di vapore alla
temperatura data esiste solo la fase vapore e lunica radice a cui si pu associare un
significato fisico (quello appunto di volume specifico del vapore) la maggiore, essendo
lunica radice esterna alla campana di Andrews (si veda la figura per il caso a 450 [K] e 5
[bar] < 9 ) [K] 450 ( P [bar]).
44
Si pu dimostrare che la soluzione centrale associabile a una condizione di equilibrio instabile (in
quanto ad un aumento di pressione corrisponde un aumento del volume molare) e quindi non viene
considerata.
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
65
10
-1
10
0
10
1
-15
-10
-5
0
5
10
15
20
25
30
35
v [m
3
kmol
-1
]
P
[
b
a
r
]
620 [K] 450 [K]
a 450 [K] < T
C
e 5
[bar] < P ha senso
fisico solo la soluzione
maggiore
a 450 [K] < T
C
e 10
[bar] > P ha senso
fisico solo la
soluzione minore
a 450 [K] < T
C
e 15
[bar] > P si ha una
sola soluzione
a 620 [K] > T
C
si
ha sempre una sola
soluzione
Lunico valore di pressione per cui non una ma due radici hanno un significato fisico la
tensione di vapore (linea a tratto e punto in figura). In questo caso sia la radice minore (a
cui si associa il significato fisico di volume specifico del liquido saturo) sia quella maggiore
(a cui si associa il significato fisico di volume specifico del vapore saturo) devono essere
considerate.
Figura 17: andamento previsto dellEoS di van der Waals sul piano P - v per il n-ottano. Si
riportano due isoterme a 450 e 620 [K], oltre alla campana di Andrews (curva con tratto
pi marcato) e allandamento sperimentale nella regione bifase (linea a tratto e punto). I
punti evidenziano le radici dellequazione per tre valori della pressione: 5, 10 e 15 [bar]. A
450 [K] la tensione di vapore del n-ottano vale circa 9 [bar], mentre la temperatura critica
pari a 569,4 [K].
Appare evidente che il principale vantaggio di questa equazione rispetto a quella del gas
perfetto la sua capacit di rappresentare il comportamento volumetrico sia della fase
vapore sia della fase liquida.
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
66
La forma dellequazione cubica nel volume viene solitamente sostituita da una forma
equivalente cubica nel coefficiente di compressibilit, Z = Pv/RT, che pu essere facilmente
dedotta dalla relazione (154):
( ) 0 1
2 3
= + + AB AZ Z B Z (155)
dove A = aP/(RT)
2
e B = bP/(RT). Le stesse considerazioni fatte per le radici
dellequazione (154) possono essere svolte per le radici dellequazione (155). Anche in
questo caso, fissate temperatura e pressione, si pu avere una sola radice reale positiva
oppure tre radici reali positive. In questultimo caso ha senso fisico il valore minore se la
pressione superiore alla tensione di vapore calcolata alla temperatura in esame
(coefficiente di compressibilit del liquido), il valore maggiore se la pressione inferiore
alla tensione di vapore (coefficiente di compressibilit del vapore) o i due valori estremi se
la pressione uguale alla tensione di vapore (coefficiente di compressibilit del vapore
saturo il maggiore e del liquido saturo il minore).
Lequazione di van der Waals non in grado di rappresentare il comportamento
volumetrico dei fluidi con unaccuratezza sufficiente per valutazioni di tipo ingegneristico.
Sono state quindi sviluppate delle varianti in grado di prevedere meglio il comportamento
sperimentale. Queste equazioni, molto utilizzate nellambito dellingegneria chimica,
mantengono la forma cubica nel volume dellequazione di van der Waals e vanno sotto il
nome di equazioni di stato cubiche. La loro forma generale la seguente:
2 2
wb ubv v
a
b v
RT
P
+ +
= (156)
che, nella forma in Z, diventa:
0
2 3
= + + + Z Z Z (157)
coi tre coefficienti della cubica che assumono le seguenti espressioni:
3 2
2 2
1
wB wB AB
uB uB wB A
uB B
=
+ =
+ =
(158)
Si noti che per u = w = 0 ci si riconduce allequazione di van der Waals. I valori di u e w
per le equazioni di stato pi utilizzate sono riportati in Tabella 5.
Come noto, possibile risolvere analiticamente unequazione cubica. In particolare,
lequazione generale (157) pu essere ricondotta alla forma normale:
0
3
= + + q pX X (159)
con la sostituzione di variabili:
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
67
+ =
=
=
3 27
2
3
3
3
2
q
p
X Z
(160)
equazione acronimo u w
van der Waals vdW 0 0 -1-B A -AB
Redlich Kwong
45
RK 1 0 -1 A-B-B
2
-AB
Redlich - Kwong
Soave
46
RKS 1 0 -1 A-B-B
2
-AB
Peng Robinson
47
PR 2 -1 -1+B A-2B-3B
2
-AB+ B
2
+ B
3
Tabella 5: valori da utilizzare nelle relazioni generali (156) e (157) per le equazioni di
stato cubiche pi note.
A seconda del segno del discriminante:
27 4
3 2
p q
D + = (161)
si hanno diverse situazioni. Se D > 0 si ha una sola soluzione reale:
3 2 2
3 / 1 3 / 1
|
.
|
\
|
+ |
.
|
\
|
+ = D
q
D
q
Z (162)
Se D = 0 si hanno tre soluzioni reali, di cui due coincidenti:
3 2
3 2
2
3 / 1
3 2
3 / 1
1
|
.
|
\
|
= =
|
.
|
\
|
=
q
Z Z
q
Z
(163)
45
O. Redlich e J.N.S. Kwong, Chem. Rev., 44, 233 (1949).
46
G. Soave, Chem. Engg. Scie., 27, 1197 (1972).
47
D.Y. Peng e D.B. Robinson, Ind. Eng. Chem. Fund., 15, 59 (1976).
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
68
Se D < 0 si hanno tre soluzioni reali distinte:
3 3
4
cos 2
3 3
2
cos 2
3 3
cos 2
3 / 1
3
3 / 1
2
3 / 1
1
|
.
|
\
| +
=
|
.
|
\
| +
=
|
.
|
\
|
=
r Z
r Z
r Z
(164)
dove:
( )
r
q
p
r
2
cos
27
3
=
=
(165)
Come detto in precedenza, i valori dei parametri a e b delle diverse equazioni di stato
devono essere stimati confrontando le previsioni dellequazione di stato con dati
sperimentali P - v - T . In alternativa, possibile ottenere una stima di tali valori imponendo
che lisoterma critica presenti un flesso orizzontale nel punto critico. Si ottiene cos un
sistema di tre equazioni nelle cinque incognite a, b, T
C
, P
C
, v
C
che per il caso dellequazione
di van der Waals assume la forma:
( )
( )
2
4 3
,
2
2
3 2
,
0
6 2
0
2
C
C
c c
C
c c
v
a
b v
RT
P
v
a
b v
RT
v
P
v
a
b v
RT
v
P
C
C
C
C
C
P T
C
C
P T
=
=
=
|
|
.
|
\
|
= +
= |
.
|
\
|
(166)
Queste tre equazioni possono essere risolte per esprimere a, b e v
C
in funzione di T
C
e P
C
.
In particolare si ottiene:
( )
C
C
C
C
P
RT
b
P
RT
a
8
64
27
2
=
=
(167)
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
69
che consentono il calcolo dei parametri a e b di qualsiasi composto una volta note
temperatura e pressione critica.
Procedendo in maniera analoga si trovano le relazioni per il calcolo dei parametri a e b
dellequazione RK, come riassunto in Tabella 6.
Le equazioni RKS e PR rappresentano degli affinamenti dellequazione RK attraverso
lintroduzione di un nuovo parametro che indica lentit della deviazione del potenziale
intermolecolare di una molecola da quello di una molecola sferica. Tale parametro, detto
fattore acentrico di Pitzer
48
, viene definito in termini di variabili ridotte, cio del valore di
una variabile rapportata al suo valore nel punto critico (in particolare, T
R
= T/T
C
e P
R
=
P/P
C
), come:
( ) 1 7 , 0 log
10
= =
R
o
R
T P (168)
dove ( ) 7 , 0 =
R
o
R
T P la tensione di vapore ridotta calcolata a una temperatura ridotta pari a
0,7. Questa definizione rende uguale a zero il valore del fattore acentrico per i fluidi
semplici (le cui molecole sono cio sferiche): argon, kripton e xenon. I valori del fattore
acentrico di Pitzer sono disponibili nei manuali tecnici
49
, e sono riportati per alcuni
composti in Tabella 4.
EoS a b s
1
s
2
s
3
vdW
( )
C
C
P
RT
2
421875 , 0
C
C
P
RT 125 , 0
0 0 0
RK
( )
R C
C
T P
RT
2
42748 , 0
C
C
P
RT 08664 , 0
0 0 0
RKS
( )
C
C
P
k RT
2
42748 , 0
C
C
P
RT 08664 , 0
0,48 1,574 -0,176
PR
( )
C
C
P
k RT
2
45724 , 0
C
C
P
RT 07780 , 0
0,37464 1,54226 -0,26992
Tabella 6: valori dei parametri a e b per le EoS cubiche pi note. Il parametro a di due EoS
(RKS e PR) dipende dalla temperatura attraverso un nuovo parametro k definito come
( ) ( )
2
1 1
R
T S k + = in cui T
R
= T/T
C
e
2
3 2 1
s s s S + + = . Il parametro detto
fattore acentrico di Pitzer.
48
K.S. Pitzer, J. Chem. Phys., 7, 583 (1939).
49
Si veda per esempio il manuale dellingegnere chimico: R.H. Perry, D.W. Green, Perrys Chemical
Engineers Handbook, McGraw-Hill, NY (1999).
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
70
LEoS RKS viene estesamente usata per il calcolo delle propriet volumetriche di
idrocarburi, mentre la PR per idrocarburi e gas inorganici come azoto e ossigeno.
3.3.2 Equazione degli stati corrispondenti
Inserendo le espressioni dei parametri a e b (167) nellequazione VdW (152) dopo aver
moltiplicato entrambi i membri per RT v e riarrangiando la relazione risultante si ottiene la
seguente equazione:
1
8
1
64
27
2
=
|
|
.
|
\
|
|
|
.
|
\
|
+
R
R
R
R
ZT
P
ZT
P
Z (169)
in cui compaiono solo, oltre al coefficiente di compressibilit, la temperatura e la pressione
ridotta. Questa equazione valida per tutti i fluidi che seguono lequazione di van der
Waals
50
e implica che il fattore di compressibilit di tutti i fluidi dipende solo dalla
temperatura e dalla pressione ridotta:
( )
R R
P T Z Z ,
0
= (170)
Questa relazione in pratica afferma che due fluidi che si trovano in due stati corrispondenti
(cio caratterizzati dalla stessa temperatura e pressione ridotta) devono avere lo stesso
coefficiente di compressibilit e rappresenta lequazione degli stati corrispondenti a due
parametri.
Nonostante lequazione di van der Waals non sia molto accurata, il comportamento
qualitativo previsto dalla relazione precedente ben verificato sperimentalmente. In altri
termini, si riscontra sperimentalmente che il fattore di compressibilit di molte specie
dipende, in prima approssimazione, solo dalle variabili ridotte, anche se la sua dipendenza
funzionale non quella prevista dallequazione (169).
Sono stati fatti molti tentativi di sviluppare delle correlazioni generali del coefficiente di
compressibilit in funzione delle variabili ridotte in grado di riprodurre correttamente le
evidenze sperimentali. La complessit delle relazioni trovate rende preferibile riportare tali
dati in forma tabulare o grafica. Tra le varie forme proposte si riportano in Appendice C e
nella Figura 18 quelle proposte da Lee e Kesler
51
.
Si nota come il fattore di compressibilit pu assumere valori anche superiori a uno, e come
in corrispondenza della tensione di vapore ridotta ad una certa temperatura ridotta si hanno
50
Si presti attenzione al fatto che questa relazione sempre lEoS vdW, semplicemente riarrangiata in
altra forma. Lunica informazione qualitativa che ci fornisce che Z appare essere funzione solo delle
variabili ridotte. Landamento corretto di Z in funzione delle variabili ridotte viene desunto da
informazioni sperimentali ed riassunto in grafici o tabelle, come discusso di seguito.
51
B.I. Lee e M.G. Kesler, AIChE J., 21, 510 (1975).
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
71
due valori del coefficiente di compressibilit, uno relativo al vapore saturo e laltro relativo
al liquido saturo.
Eccetto che nella regione prossima alla curva di saturazione e al punto critico questa
correlazione prevede il coefficiente di compressibilit di composti poco polari con un errore
di circa il 5 %. Daltro canto, per composti molto polari e per i cosiddetti gas quantici
(idrogeno, elio e neon) gli scostamenti dai valori sperimentali possono essere molto elevati.
Per migliorare la previsione dellequazione degli stati corrispondenti per i composti polari
si introdotto un terzo parametro che tiene conto della polarit delle molecole. Dalla Figura
18 si nota come lequazione degli stati corrispondenti a due parametri prevede che il
coefficiente di compressibilit nel punto critico sia uguale a 0,27. In realt, come si pu
vedere anche dalla Tabella 4, il valore sperimentale varia solitamente tra 0,23 e 0,31.
quindi possibile utilizzare tale valore come terzo parametro.
Figura 18: fattore di compressibilit in funzione di temperatura e pressione ridotta nella
formulazione di Lee - Kesler.
Pi comune per lapproccio di Lee - Kesler che utilizza il fattore acentrico di Pitzer
come terzo parametro, modificando quindi la relazione (170) nella seguente:
10
-2
10
-1
10
0
10
1
0
0.2
0.4
0.6
0.8
1
P
r
[-]
Z
zona
bifase
liquido
gas
vapore
saturo
liquido
saturo
Tr=4
Tr=0,7
Tr=0,9
Tr=1
Tr=1,5
punto
critico
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
72
( ) ( ) ( ) ( ) ( )
R R R R R R R R
P T Z P T Z
Z
P T Z P T Z Z , , 0 , , ,
1 0
0
0
+ = |
.
|
\
|
+ =
=
(171)
In questa relazione (detta equazione degli stati corrispondenti a tre parametri) si
approssimata la dipendenza di Z dal fattore acentrico con unespansione in serie di Taylor
troncata al primo termine nellintorno del valore = 0 e si indicato con Z
1
il valore della
derivata di Z rispetto a valutata in = 0
52
. Il valore di Z
1
al variare di temperatura e
pressione ridotta riportato in Appendice C. Esso rappresenta una correzione al valore di Z
0
e pu quindi essere spesso trascurato per una valutazione di massima.
3.3.3 Equazione del viriale
Nel 1901 Onnes
53
propose la seguente equazione di stato per i composti gassosi:
...
) ( ) (
1
2
+ + + = =
v
T C
v
T B
RT
Pv
Z (172)
dove B, C sono parametri caratteristici del tipo di composto considerato. Si pu
dimostrare
54
che tali parametri sono dipendenti solo dalla temperatura e rappresentano le
interazioni tra due molecole (il parametro B) e tra tre molecole (il parametro C). Essi
vengono chiamati secondo e terzo coefficiente del viriale e i loro valori sono stati stimati
per un gran numero di composti
55
.
Lequazione precedente pu anche essere scritta in serie di potenze della pressione:
... ) ( ' ) ( ' 1
2
+ + + = = P T C P T B
RT
Pv
Z (173)
dove B = B/(RT) e C = (C-B
2
)/(RT)
2
.
Il principale vantaggio dellequazione del viriale che pu essere resa sempre pi precisa
aumentando il numero di termini. Per valutazioni ingegneristiche, la forma troncata al
secondo coefficiente fornisce solitamente prestazioni ragionevoli per pressioni non troppo
elevate (inferiori a circa 15 [bar], o equivalentemente per valori della densit ridotta
5 , 0 25 , 0 < =
C R
).
52
In altri termini, si linearizzata la dipendenza di Z da nellintorno di = 0.
53
Heike Kamerlingh Onnes, 1853 1926, fisico olandese.
54
Lequazione di stato del viriale pu essere dedotta applicando la meccanica statistica e quindi ha
delle basi teoriche pi solide di altre equazioni di stato semiempiriche. Si veda per esempio: T. L. Hill
Introduzione alla termodinamica statistica, Piccin, Padova, 1970.
55
Si veda per esempio J.H. Dymond e E.B. Smith, The virial coefficients of pure gases and mixtures,
Clarendon Press, Oxford, 1980.
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
73
In queste condizioni la relazione precedente diventa:
( ) , , 1 1 ) ( ' 1
R R
R
R
C
C
P T Z
T
P
RT
BP
RT
BP
P T B Z =
|
|
.
|
\
|
+ = + = + (174)
Si pu quindi esprimere il termine tra parentesi con una relazione analoga alla (171):
( ) ( )
R R
C
C
T B T B
RT
BP
1 0
+ =
(175)
dove si persa la dipendenza dalla pressione in quanto il secondo coefficiente del viriale
dipende solo dalla temperatura. I parametri B
0
e B
1
possono essere approssimati con le
relazioni:
( )
( )
2 , 4
1
6 , 1
0
172 , 0
139 , 0
422 , 0
083 , 0
R
R
T
T B
T
T B
R
R
=
=
(176)
Queste relazioni forniscono risultati simili alle relazioni di Lee - Kesler a qualsiasi
pressione per T
R
> 3. A temperature inferiori lintervallo di pressione in cui le relazioni
forniscono valori accettabili diminuisce. A T
R
= 2 il valore limite circa P
R
= 2, mentre a
T
R
= 0,7 il valore limite pari a quello di saturazione.
La principale limitazione dellequazione del viriale la sua capacit di rappresentare
ragionevolmente bene solo il comportamento della fase gas. In altri termini, non in grado
di prevedere la transizione di fase da gas a liquido.
3.4 Applicazione
Si calcoli il volume molare del n-ottano in fase vapore a 427.9 [K] e 2.15 [bar] con le
diverse equazioni di stato viste in questo capitolo.
Gas ideale
] mol [m 0165 , 0
[Pa] 10 15 , 2
[K] 9 , 427 ] K mol [J 314 , 8
1 - 3
5
1 1
=
= =
P
RT
v (177)
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
74
vdW
( ) ( )
] mol m [J 7863 , 3
10 97 , 24
40 , 569 314 , 8 421875 , 0 421875 , 0
2 3
5
2 2
= =
C
C
P
RT
a
] [m 0002 , 0
10 97 , 24
40 , 569 314 , 8 125 , 0 125 , 0
3
5
=
= =
C
C
P
RT
b
(178)
( )
0643 , 0
2
= =
RT
aP
A
0143 , 0 = =
RT
bP
B
(179)
-1,0143 1 = = B
0,0643 = = A
-0,0009 = = AB
(180)
-0,2786
3
2
= =
p
-0,0565
3 27
2
3
= + =
q
6
3 2
10 -3,7556
27 4
= + =
p q
D
(181)
Poich D < 0 lequazione ammette tre soluzioni reali distinte, che si possono calcolare
come segue.
I valori dei parametri necessari al calcolo delle radici sono:
0,0283
27
3
= =
p
r
[rad] 0,0685
2
cos
1
= |
.
|
\
|
=
r
q
(182)
I valori delle tre radici sono i seguenti:
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
75
0,0455
3 3
4
cos 2
0,0214
3 3
2
cos 2
0,9475
3 3
cos 2
3 / 1
3
3 / 1
2
3 / 1
1
= |
.
|
\
| +
=
= |
.
|
\
| +
=
= |
.
|
\
|
=
r Z
r Z
r Z
(183)
Poich si ricerca il volume molare del gas, la radice maggiore quella da utilizzare, da cui
si ricava:
] mol [m 0157 , 0
1 - 3
1
= =
P
RT
Z v (184)
RK
( )
] mol m [J 4,4258
42748 , 0
2 3
2
= =
R C
C
T P
RT
a
] [m 10 6426 , 1
08664 , 0
3 4
= =
C
C
P
RT
b
(185)
( )
0752 , 0
2
= =
RT
aP
A
0099 , 0 = =
RT
bP
B
(186)
-1,0000 =
0652 , 0 =
-0,0007 =
(187)
-0,2682 = p
-0,0531 = q
6
10 9660 , 9
= D
(188)
Poich D < 0 lequazione ammette tre soluzioni reali distinte, che si possono calcolare
come segue.
0,0267 = r (189)
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
76
[rad] 0,1161 =
0,8619
0,0152
0,9306
3
2
1
=
=
=
Z
Z
Z
(190)
Poich si ricerca il volume molare del gas, la radice maggiore quella da utilizzare:
] mol [m 0154 , 0
1 - 3
1
= =
P
RT
Z v (191)
RKS
0786 , 1 398 , 0 176 , 0 398 , 0 574 , 1 48 , 0 176 , 0 574 , 1 48 , 0
2 2
= + = + = S
( ) ( ) ( ) ( ) 3078 , 1 7515 , 0 1 0786 , 1 1 1 1
2 2
= + = + =
R
T S k
( )
] mol m [J 5,0174
42748 , 0
2 3
2
= =
C
C
P
k RT
a
] [m 10 6426 , 1
08664 , 0
3 4
= =
C
C
P
RT
b
(192)
( )
0852 , 0
2
= =
RT
aP
A
0099 , 0 = =
RT
bP
B
(193)
-1,0000 =
0752 , 0 =
-0,0008 =
(194)
-0,2581 = p
-0,0499 = q
5
10 7190 , 1
= D
(195)
Poich D < 0 lequazione ammette tre soluzioni reali distinte, che si possono calcolare
come segue:
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
77
0,0252 = r
[rad] 0,1577 =
(196)
0,0671
0,0137
0,9192
3
2
1
=
=
=
Z
Z
Z
(197)
Poich si ricerca il volume molare del gas, la radice maggiore quella da utilizzare:
] mol [m 0152 , 0
1 - 3
1
= =
P
RT
Z v (198)
PR
9457 , 0 26992 , 0 54226 , 1 37464 , 0
2
= + = S
( ) ( ) 2676 , 1 1 1
2
= + =
R
T S k
( )
] mol m [J 5,2020
45724 , 0
2 3
2
= =
C
C
P
k RT
a
] [m 10 474 , 1
07780 , 0
3 4
= =
C
C
P
RT
b
(199)
( )
0837 , 0
2
= =
RT
aP
A
0089 , 0 = =
RT
bP
B
(200)
-0,9911 =
0703 , 0 =
-0,0007 =
(201)
-0,2571 = p
-0,0496 = q
5
10 4663 , 1
= D
(202)
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
78
Poich D < 0 lequazione ammette tre soluzioni reali distinte, che si possono calcolare
come segue:
0,0251 = r
[rad] 0,1532 =
(203)
0,0638
0,0121
0,9151
3
2
1
=
=
=
Z
Z
Z
(204)
Poich si ricerca il volume molare del gas, la radice maggiore quella da utilizzare:
] mol [m 0151 , 0
1 - 3
1
= =
P
RT
Z v (205)
Stati corrispondenti
Dalle tabelle di Lee - Kesler a T
R
= 0,75 e P
R
= 0,086 si ricava (interpolando i valori
riportati):
Z
0
= 0,93
Z
1
= -0,06
(206)
e quindi
9 , 0
1 0
= + = Z Z Z (207)
] mol [m 0150 , 0
1 - 3
= =
P
RT
Z v (208)
Viriale
( )
( ) 4320 , 0
172 , 0
139 , 0
5834 , 0
422 , 0
083 , 0
2 , 4
1
6 , 1
0
= =
= =
R
R
T
T B
T
T B
R
R
(209)
( ) ( ) 7555 , 0
1 0
= + =
R R
C
C
T B T B
RT
BP
(210)
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
79
9134 , 0 1 =
|
|
.
|
\
|
+
R
R
C
C
T
P
RT
BP
Z (211)
] mol [m 0151 , 0
1 - 3
= =
P
RT
Z v (212)
I risultati ottenuti sono riassunti nella Tabella 7. Si nota che non vi sono grandi differenze
tra le diverse equazioni di stato in quanto il comportamento del fluido prossimo a quello
di un gas perfetto.
Seguendo lo stesso procedimento anche possibile calcolare il volume molare del n-ottano
liquido alla stessa temperatura ma a una pressione di 5 [bar]. Questa volta, come riportato
in Tabella 7, le differenze sono marcate. In particolare, le equazioni di stato del gas perfetto
e del viriale forniscono ovviamente risultati non corretti. Inoltre, anche lequazione di van
der Waals fornisce risultati diversi da quelli delle altre equazioni, che risultano viceversa
abbastanza equivalenti.
EoS v
G
(427,9 [K] e 2,15 [bar]) v
L
(427,9 [K] e 5 [bar])
Gas perfetto 0,0165 7,12 10
-3
vdW 0,0157 3,51 10
-4
RK 0,0154 2,43 10
-4
RKS 0,0152 2,26 10
-4
PR 0,0151 2,00 10
-4
Stati corrispondenti 0,0150 1,99 10
-4
Viriale 0,0151 5,68 10
-3
Tabella 7: valori dei volumi molari [m
3
mol
-1
] calcolati con diverse EoS.
Lo svolgimento di questo tipo di conti evidentemente tedioso, soprattutto se necessario
ripeterli un gran numero di volte.
Risulta quindi utile implementare la sequenza dei calcoli in un programma di calcolo. A
titolo di esempio, si riporta di seguito un programma in linguaggio MATLAB. Il programma
commentato e quindi di facile comprensione e pu essere implementato in un file testo di
nome vTP.m per essere eseguito in ambiente MATLAB. Si segnala solo che previsto il
calcolo del volume molare del vapore (variabile fase=1) o del liquido (variabile
fase=2) e che la risoluzione delle equazioni di stato cubiche demandata alla routine
esterna EoS, il cui listato riportato di seguito alla routine vTP e pu essere implementato
in un file testo di nome EoS.m. La variabile tipo definisce il tipo di equazione di stato
cubica.
Appare evidente il vantaggio di questo approccio: possibile ripetere i calcoli a diverse
pressioni o per diversi composti modificando solo i dati in testa al programma, mentre i
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
80
calcoli a diverse temperature richiedono anche la scrittura dei valori dedotti dalle tabelle di
Lee - Kesler che sono riportati solo per il valore di T
R
=0,75.
%vTP calcolo del volume molare
% a T e P con diverse EoS
% R. Rota (2001)
format short g %formato scrittura
clear all %cancella tutte le variabili
%dati per n-ottano
Tc =569.4; %[K]
Pc =24.97e5; %[Pa]
om =0.398; %fattore acentrico di Pitzer
%calcolo volume molare gas (fase=1) o liquido (fase=2)
fase = 1;
%assegno le condizioni di T e P
T = 427.9; %temperatura [K]
P = 2.15e5; %pressione [Pa]
R = 8.314; %[J/(mol K)]
RT =R*T;
RTc=R*Tc;
TR =T/Tc;
PR =P/Pc;
%1 gas perfetto
disp('gas perfetto')
v = R*T/P
%2 vdW, RK, RKS, PR
eq=['vdW';'RK ';'RKS';'PR ']; %nomi EoS
for tipo=1:4 %ciclo su diverse EoS
Z = EoS(T,P,Tc,Pc,om,tipo); %calcolo Z a (P,T)
disp(eq(tipo,:)) %visualizzo nome EoS
if fase == 1
v = Z(3)*RT/P %calcolo Vgas [m3/mol]
else
v = Z(1)*RT/P %calcolo Vliq [m3/mol]
end
end
%3 stati corrispondenti @ TR=0.75
clear Z; %cancello il vettore Z
%vettori coi valori di PR Z0 e Z1 a TR=0,75
PRZ=[.01 .05 .1 .2 .4 .6 .8 1 1.2 1.5 2 3 5 7 10];
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
81
Z0 =[.9922 .9598 .9165 .0336 .067 .1001 .133 .1656 ...
.1981 .2426 .3260 .4823 .7854 1.0787 1.5047];
Z1 =[-.0064 -.0339 -.0744 -.0143 -.0282 -.0417 -.055 ...
-.0681 -.0808 -.0996 -.1298 -.1872 -.2929 ...
-.3901 -.525];
Z0i=interp1(PRZ,Z0,PR); %interpolo Z0 al valore di PR
Z1i=interp1(PRZ,Z1,PR); %interpolo Z1 al valore di PR
Z = Z0i + om*Z1i; %calcolo Z
disp('stati corrispondenti')
v = Z*RT/P %calcolo V [m3/mol]
%4 viriale
B0 = 0.083-0.422/TR^1.6; %calcolo B0
B1 = 0.139-0.172/TR^4.2; %calcolo B1
BPcRTc = B0+om*B1; %calcolo BPc/RTc
Z = 1+BPcRTc*PR/TR; %calcolo Z
disp('viriale')
v = Z*RT/P %calcolo V [m3/mol]
function [Z,A,B,S,k] = EoS(T,P,Tc,Pc,om,tipo)
%EoS calcola Z con diverse EoS
% dati T [K], P [Pa], Tc [K],
% Pc [Pa], om [-], tipo (EoS da usare)
% tipo: 1 vdW
% 2 RK
% 3 RKS
% 4 PR
% restituisce Z (ordinato) oppure Z, A, B, S e k
%
% uso: Z = EoS(T,P,Tc,Pc,om,tipo)
% [Z,A,B,S,k] = EoS(T,P,Tc,Pc,om,tipo)
% R. Rota (2002)
R =8.314; %costante gas perfetti [J/(mol K)]
RT =R*T;
RTc=R*Tc;
%parametri dell'equazione nella forma Z^3+alfa*Z^2+beta*Z+gamma=0
if tipo == 1 %vdW
a =27*RTc^2 / (64*Pc);
b =RTc / (8*Pc);
A =a*P/(RT)^2;
B =b*P/(RT);
alfa =-1-B;
beta =A;
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
82
gamma=-A*B;
end
if tipo ==2 %RK
TR =T/Tc;
a =0.42748*RTc^2 / (Pc*sqrt(TR));
b =0.08664*RTc / Pc;
A =a*P/(RT)^2;
B =b*P/(RT);
alfa =-1;
beta =A-B-B^2;
gamma=-A*B;
end
if tipo ==3 %RKS
k =0.48+1.574*om-0.176*om^2;
alT=(1+k*(1-sqrt(T/Tc)))^2;
a =0.42748 * alT * RTc^2 / Pc;
b =0.08664 * RTc / Pc;
A =a*P/(RT)^2;
B =b*P/(RT);
alfa =-1;
beta =A-B-B^2;
gamma=-A*B;
end
if tipo == 4 %PR
k =0.37464+1.54226*om-0.26992*om^2;
alT=(1+k*(1-(T/Tc)^0.5))^2;
a =0.45724 * alT * RTc^2 / Pc;
b =0.07780 * RTc / Pc;
A =a*P/(RT)^2;
B =b*P/(RT);
alfa =-1+B;
beta =A-2*B-3*B^2;
gamma=-A*B+B^2+B^3;
end
%risoluzione analitica della cubica
p = beta-alfa^2/3;
q = 2*alfa^3/27-alfa*beta/3+gamma;
q2=q/2;
a3=alfa/3;
D = q^2/4+p^3/27;
if D > 0
Z1 = (-q2+sqrt(D))^(1/3)+(-q2-sqrt(D))^(1/3)-a3;
Z = [Z1 Z1 Z1];
elseif D == 0
Z1 = -2*(q2)^(1/3)-a3;
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
83
Z2 = (q2)^(1/3)-a3;
Z = [Z1 Z2 Z2];
else
r = sqrt(-p^3/27);
teta= acos(-q2*sqrt(-27/p^3));
Z1 = 2*r^(1/3)*cos(teta/3)-a3;
Z2 = 2*r^(1/3)*cos((2*pi+teta)/3)-a3;
Z3 = 2*r^(1/3)*cos((4*pi+teta)/3)-a3;
Z = [Z1 Z2 Z3];
end
Z =sort(Z); %ordina i valori
Si noti che la risoluzione analitica della cubica pu essere sostituita dalla ricerca numerica
delle radici di un polinomio effettuata dalla funzione roots di MATLAB. Lultima parte
della routine EoS pu quindi essere sostituita dai seguenti comandi:
%risoluzione numerica della cubica
coeff =[ 1
alfa
beta
gamma];
Z = roots(coeff); %calcola le radici di un polinomio
Z =sort(Z); %ordina i valori
Un programma analogo pu essere utilizzato per visualizzare le previsioni delle diverse
equazioni di stato. Il programma riportato di seguito (che pu essere implementato in un
file testo di nome Pv.m e quindi eseguito in ambiente MATLAB) traccia su di un piano P - v
lisoterma a 450 [K] calcolata con diverse equazioni di stato per il n-ottano. Il risultato,
riportato in Figura 19, mostra come quando il comportamento del fluido approssima quello
di un gas perfetto (per bassi valori della pressione) tutte le equazioni forniscono risultati
analoghi. Lequazione del gas perfetto si scosta poi per prima dalle altre man mano che ci si
avvicina al vapore saturo. Lequazione di stato del viriale non in grado di prevedere la
transizione di fase (regione con radici multiple delle equazioni cubiche), mentre anche le
previsioni delle diverse equazioni di stato cubiche differiscono significativamente nella
regione del liquido. Questo comportamento abbastanza generale: le equazioni di stato,
nascendo come correzioni al comportamento di gas perfetto, prevedono generalmente
meglio il comportamento dei gas rispetto a quello dei liquidi.
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
84
Figura 19: isoterma a 450 [K] per il n-ottano calcolata con diverse EoS.
%Pv disegno delle isoterme con diverse EoS cubiche
% tipo = 1 vdW
% 2 RK
% 3 RKS
% 4 PR
% 5 gas perfetto
% 6 viriale
% R. Rota (2001)
format short g %scrittura a video compatta
clear all %cancella tutte le variabili
close %chiude le finestre grafiche
%dati per n-ottano
Tc =569.4; %[K]
Pc =24.97e5; %[Pa]
om =0.398; %fattore acentrico di Pitzer
10
-1
10
0
10
1
-80
-60
-40
-20
0
20
40
v [m
3
kmol
-1
]
P
[
b
a
r
]
gas perfetto
viriale
vdW RK
RKS
PR
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
85
%tracciamento delle isoterme
T = 450; %[K]
R = 8.314; %[J/(mol K)]
RT =R*T;
RTc=R*Tc;
TR =T/Tc;
Vj=logspace(log10(2.4e-4),-2,1000); %spaziatura log.
for tipo=1:6
Vau = []; Pau = [];
for j=1:length(Vj)
V=Vj(j);
if tipo == 1
a =27*RTc^2 / (64*Pc);
b =RTc / (8*Pc);
P = RT/(V-b)-a/V^2; %vdW
end
if tipo == 2
a =0.42748*RTc^2 / (Pc*sqrt(TR));
b =0.08664*RTc / Pc;
P = RT/(V-b)-a/(V*(V+b)); %RK
end
if tipo == 3
Vj=logspace(log10(2.25e-4),-2,1000);%spaziatura
k =0.48+1.574*om-0.176*om^2;
alT=(1+k*(1-sqrt(T/Tc)))^2;
a =0.42748 * alT * RTc^2 / Pc;
b =0.08664 * RTc / Pc;
P = RT/(V-b)-a/(V*(V+b)); %RKS
end
if tipo == 4
Vj=logspace(log10(2.e-4),-2,2000); %spaziatura
k =0.37464+1.54226*om-0.26992*om^2;
alT=(1+k*(1-(T/Tc)^0.5))^2;
a =0.45724 * alT * RTc^2 / Pc;
b =0.07780 * RTc / Pc;
P = RT/(V-b)-a/(V*(V+b)+b*(V-b)); %PR
end
if tipo == 5
P = RT/V; %gas perfetto
end
if tipo == 6
B0 = 0.083-0.422/TR^1.6;
B1 = 0.139-0.172/TR^4.2;
BPcRTc = B0+om*B1;
P = 1/(V/RT-BPcRTc*Tc/(Pc*T)); %viriale
end
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
86
Vau = [Vau; V]; %memorizzo il valore di V
Pau = [Pau; P]; %memorizzo il valore di P
end
semilogx(Vau*1e3,Pau*1e-5,'k-')
hold on
clear Vau Pau
end
xlabel('v [m^3 kmol^{-1}]')
ylabel('P [bar]')
axis([1e-1 1e1 -80 50])
semilogx([1e-1 1e1],[0 0],'k-')
text(1,40,'gas perfetto')
text(1,20,'viriale')
text(.4,10,'vdW')
text(.26,10,'RK')
text(.27,-50,'RKS')
text(.2,-70,'PR')
3.5 Esercizi
Calcolare il volume specifico di una miscela in equilibrio liquido - vapore di acqua a titolo
0,8 alla temperatura di 150 [C] utilizzando le tabelle termodinamiche dellacqua.
Risultato: 0,314 [m
3
kg
-1
].
La tensione di vapore dellacqua a 180 [C] pari a 10,027 [bar]. Calcolare il fattore
acentrico dellacqua.
Risultato: 0,333.
Un serbatoio da 1 [m
3
] contiene 5 [kg] di ammoniaca a 300 [K]. Calcolare la pressione nel
serbatoio utilizzando sia lequazione di stato del gas perfetto, sia quella di van der Waals.
Risultato: 7,3 [bar]; 7,1 [bar].
Si vuole stoccare dellammoniaca in un serbatoio di 10 [m
3
] a 321,55 [K] e 19,5 [bar]. Se
per ragioni di sicurezza il volume di liquido non deve eccedere il 70% del volume del
serbatoio, calcolare la massima quantit di ammoniaca che pu essere immagazzinata nel
serbatoio utilizzando lequazione di stato RK.
Risultato: 2964 [kg].
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
87
Calcolare il volume di un serbatoio necessario per stoccare 100 [kg] di metano a 25 [C] e
20 [bar] con lequazione di stato RKS.
Risultato: 7,5 [m
3
].
Calcolare il volume di un serbatoio necessario per stoccare 1000 [kg] di etano a 400 [K] e
10 [bar] con lequazione di stato PR.
Risultato: 107,1 [m
3
].
Calcolare il volume di un serbatoio necessario per stoccare 500 [kg] di etilene a 25 [C] e
10 [bar] con lequazione di stato del viriale.
Risultato: 41,8 [m
3
].
Un serbatoio da 1 [m
3
] contenente 180 [kg] di anidride carbonica viene posizionato in
prossimit di un forno. Calcolare la pressione nel serbatoio quando la sua temperatura
raggiunge i 91,8 [C] utilizzando lequazione degli stati corrispondenti a due parametri.
Risultato: 90 [bar].
Si calcoli la pressione generata da 1 [lb mol] di metano confinata in un recipiente da 2 [ft
3
]
a 122 [F] utilizzando la legge degli stati corrispondenti a due parametri.
Risultato: 190 [bar].
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
89
4 Propriet termodinamiche dei fluidi puri
Nei capitoli precedenti sono state introdotte delle grandezze termodinamiche non misurabili
sperimentalmente (come lenergia interna, lentalpia e lentropia) per caratterizzare lo stato
di un sistema. Nel caso di un gas perfetto, si visto che semplici relazioni consentono il
calcolo di queste grandezze dai valori delle grandezze misurabili (temperatura, pressione e
volume specifico). Daltro canto, nella pratica quotidiana lingegnere chimico deve
eseguire dei calcoli che coinvolgono fluidi il cui comportamento volumetrico non
assimilabile a quello di un gas perfetto
56
. Lo scopo principale di questo capitolo quindi
quello di dedurre delle relazioni per il calcolo delle grandezze termodinamiche non
misurabili dei fluidi puri. Tutta la parte iniziale del capitolo relativa a sistemi omogenei;
lestensione a sistemi eterogenei, trattandosi di variabili estensive, ne deriva naturalmente
come somma del valore delle variabili di ciascuna fase. Per ragioni di generalit che
torneranno utili nel prosieguo del testo, le relazioni fondamentali sviluppate allinizio del
capitolo vengono presentate per sistemi multicomponente, da cui le relazioni per sistemi
monocomponente possono essere dedotte come caso particolare.
Al termine dello studio di questo capitolo lo studente dovrebbe conoscere:
lenergia libera di Helmholtz e di Gibbs e la loro origine;
le relazioni di Maxwell;
il principio della minima energia e le condizioni di equilibrio in sistemi soggetti a
diversi vincoli;
il concetto di funzione residua;
le relazioni tra lenergia libera di Gibbs residua e le altre funzioni residue;
come derivare la relazione generale per il calcolo dellenergia libera di Gibbs residua
utilizzando una equazione di stato;
come dedurre le relazioni analitiche per il calcolo dellentalpia e dellentropia residua
con lEoS del viriale;
come calcolare lentalpia e lentropia residua di liquidi e vapori con EoS cubiche;
come calcolare lentalpia e lentropia residua di liquidi e vapori con lequazione degli
stati corrispondenti;
come calcolare entalpia ed entropia di un fluido ad assegnata temperatura e pressione;
come calcolare la variazione di entalpia ed entropia di un fluido tra due stati;
come calcolare entalpia ed entropia di un fluido ad assegnata temperatura e pressione
coi diagrammi di stato o le tabelle termodinamiche;
come calcolare la variazione di entalpia ed entropia di un fluido tra due stati, di cui uno
condensato, utilizzando i calori latenti di transizione di fase;
il coefficiente di Joule - Thomson;
56
Basta pensare ai liquidi.
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
90
lequazione di Clapeyron, di Clausius - Clapeyron e di Antoine;
il concetto di fugacit;
come calcolare la fugacit di un fluido puro ad assegnata temperatura e pressione con
le diverse EoS;
come calcolare la fugacit di un fluido puro in una fase condensata utilizzando la
tensione di vapore;
la correzione di Poynting;
come calcolare la tensione di vapore utilizzando unequazione di stato;
come calcolare la fugacit di un solido utilizzando il calore latente di fusione.
4.1 Funzioni termodinamiche
Lo stato di equilibrio di un sistema completamente caratterizzato dallequazione
fondamentale (14)
57
:
) , , ( n V U S S = (213)
Questa relazione funzionale pu essere risolta rispetto ad U per fornire la relazione
equivalente
58
:
) , , ( n V S U U = (214)
Queste due funzioni sono equivalenti nel senso che entrambe forniscono tutte le
informazioni termodinamiche sullo stato del sistema in equilibrio. In particolare, attraverso
le relazioni (16) possibile calcolare la temperatura e la pressione del sistema come
derivate di S oppure di U. Questo conduce alla relazione (19) per un sistema omogeneo:
=
+ =
N
i
i
i
dn
T
dV
T
P
dU
T
dS
1
1
(215)
o alla sua forma equivalente:
=
+ =
N
i
i i
dn PdV TdS dU
1
(216)
Per un fluido puro queste due relazioni diventano:
57
Si visto come lentropia consente di definire la direzione in cui evolve spontaneamente un sistema
isolato (cio un qualunque sistema pi il suo ambiente) e quindi il suo stato di equilibrio.
58
In quanto lentropia una funzione continua e differenziabile monotonamente crescente con
lenergia interna.
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
91
dn
T
dV
T
P
dU
T
dS
+ =
1
(217)
dn PdV TdS dU + = (218)
che, come gi visto, si semplificano ulteriormente nel caso di un sistema chiuso (per cui
dn=0):
dV
T
P
dU
T
dS + =
1
(219)
PdV TdS dU = (220)
Il senso fisico di queste relazioni (prendendo come esempio la equazione (216)) che la
variazione di una grandezza estensiva (U) viene calcolata sulla base delle variazioni di altre
grandezze estensive (S, V e n). Purtroppo, una di queste variabili, S, non misurabile.
Questo un problema per un ingegnere che sarebbe molto pi interessato a una relazione in
grado di fornire la variazione di U sulla base delle variazioni di grandezze misurabili (T, P,
V o n); ci renderebbe possibile il calcolo della variazione di U sulla base di misure
sperimentali. In pratica, per ottenere una relazione utile per un ingegnere necessario
sostituire alla variabile estensiva S nella relazione (216) una variabile misurabile, per
esempio T, cos da ottenere una relazione funzionale del tipo:
) , , ( n V T U U = (221)
da cui dedurre una nuova espressione per il calcolo della variazione di energia interna:
=
=
+ =
=
|
|
.
|
\
|
+ |
.
|
\
|
+ |
.
|
\
|
N
i
i i V
N
i
i
n V T
i T V
dn PdV dT C
dn
n
U
dV
V
U
dT
T
U
dU
i j
1
1
, ,
, ,
n n
(222)
Questo in linea di principio possibile poich esiste un legame tra S, U e T dato dalla
relazione (16):
T
S
U
V
= |
.
|
\
|
n ,
(223)
Il problema che questo cambiamento di variabili comporta una perdita di informazioni
sullo stato del sistema. Infatti, si sostituisce alla variazione di una funzione (dS) la
variazione di una sua derivata (dT), cio la variazione della tangente alla curva S=S(U)
V,n
.
La perdita di informazioni in questo passaggio evidente se si considera che pu esistere
unintera famiglia di curve S(U) caratterizzate dalla stessa tangente: noto il valore della
tangente, non possibile risalire in modo univoco alla curva originale.
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
92
Si pu dimostrare che possibile sostituire in una relazione funzionale tipo la (221) la
dipendenza da una variabile estensiva con quella dalla derivata della funzione rispetto alla
variabile estensiva che si vuole eliminare senza perdere alcuna informazione se si applica la
cosiddetta trasformata parziale di Legendre
59
. Data una funzione y delle variabili x:
) ,..., , (
2 1 n
x x x y y = (224)
la trasformata parziale di Legendre genera una nuova funzione in cui la dipendenza da
alcune variabili x
j
viene sostituita dalla dipendenza dalla derivata ad essa correlata,
( )
j i
x
j j
x y m
= , definita come
60
:
= =
n
j k
k k n j j
x m y m m x x y y ) ,..., , ,..., (
1 1
( (
(225)
Partendo dalla relazione fondamentale (221), si possono dedurre delle nuove funzioni
termodinamiche con lo stesso contenuto informativo, in grado quindi, per esempio, di
definire la direzione delle trasformazioni spontanee e lo stato di equilibrio. Le funzioni
termodinamiche pi utilizzate nella pratica ingegneristica sono descritte di seguito.
4.1.1 Entalpia
Sostituendo nella relazione (221) al volume la derivata ad esso correlata,
( ) P V U m
, S
= =
n 2
, si ottiene la nuova funzione
) , , ( ) , , ( n n P S H PV U P S U U = + = =
( (
(226)
Lentalpia, H, che era stata introdotta in precedenza per motivi di praticit, risulta quindi
essere una funzione che conserva il contenuto informativo delle relazioni fondamentali.
Sfortunatamente, essa dipende ancora da una variabile non misurabile, lentropia.
Il suo differenziale primo infatti :
( )
=
=
+ + =
= + + + = + =
N
i
i i
N
i
i i
dn VdP TdS
VdP PdV dn PdV TdS PV d dU dH
1
1
(227)
che, come atteso, dipende solo dalla variazione delle grandezze S, P e n.
59
Adrien Marie Legendre, 1752?-1833, matematico francese
60
Si veda per esempio H.B. Callen Thermodynamics, John Wiley & Sons, N.Y. (1960) per
maggiori dettagli.
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
93
Nel caso di un fluido puro:
dn VdP TdS dH + + = (228)
Se si considera poi anche un sistema chiuso:
VdP TdS dH + = (229)
In termini specifici, la relazione per un composto puro diventa ovviamente:
vdP Tds dh + = (230)
4.1.2 Energia libera di Helmholtz
61
Sostituendo nella relazione (221) allentropia la derivata ad esso correlata,
( ) T S U m
, V
= =
n 1
, si ottiene la nuova funzione
) , , ( ) , , ( n n V T A TS U V T U U = = =
( (
(231)
Lenergia libera di Helmholtz, A, dipende solo da variabili misurabili e, ovviamente,
conserva il contenuto informativo delle relazioni fondamentali.
Il suo differenziale primo :
( )
=
=
+ =
= + = =
N
i
i i
N
i
i i
dn PdV SdT
SdT TdS dn PdV TdS TS d dU dA
1
1
(232)
che dipende solo dalla variazione delle grandezze T, V e n.
Nel caso di un fluido puro:
dn PdV SdT dA + = (233)
Se si considera poi anche un sistema chiuso:
PdV SdT dA = (234)
In termini specifici, la relazione per un composto puro diventa ovviamente:
Pdv sdT da = (235)
61
Hermann Ludwig Ferdinand Helmholtz, 1821 - 1894, fisico tedesco.
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
94
4.1.3 Energia libera di Gibbs
62
Nonostante lenergia libera di Helmholtz dipenda solo dalle variabili misurabili T, V e n,
queste non sono le pi comode per effettuare dei conti ingegneristici. Infatti, le variabili che
vengono controllate e misurate in un impianto sono solitamente temperatura e pressione
(oltre alla quantit di materia).
Sostituendo nella relazione (221) allentropia la derivata ad esso correlata,
( ) T S U m
V
= =
n , 1
, e al volume la derivata ad esso correlata, ( ) P V U m
S
= =
n , 2
, si
ottiene la nuova funzione
) , , ( ) , , ( n n P T G TS H PV TS U P T U U = = + = =
( (
(236)
Lenergia libera di Gibbs, G, dipende solo da variabili misurabili e conserva il contenuto
informativo delle relazioni fondamentali.
Il suo differenziale primo :
( )
=
=
+ + =
= + + + =
= + =
N
i
i i
N
i
i i
dn VdP SdT
VdP PdV SdT TdS dn PdV TdS
PV d TS d dU dG
1
1
) (
(237)
che dipende solo dalla variazione delle grandezze T, P e n.
Nel caso di un fluido puro:
dn VdP SdT dG + + = (238)
Se si considera poi anche un sistema chiuso:
VdP SdT dG + = (239)
In termini specifici, la relazione per un composto puro diventa ovviamente:
vdP sdT dg + = (240)
Lenergia libera di Gibbs, come verr estesamente illustrato nel seguito, gioca un ruolo
assolutamente fondamentale nella termodinamica dellingegneria chimica.
62
Josiah Willard Gibbs, 1839 - 1903, fisico matematico statunitense.
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
95
4.1.4 Relazioni di Maxwell
63
Un aspetto formale della termodinamica rappresentato dalle cosiddette relazioni di
Maxwell. Si tratta di un insieme di relazioni matematiche tra le derivate delle grandezze
termodinamiche che nasce dal fatto che il differenziale delle funzioni di stato esatto e che
quindi i coefficienti dei differenziali delle variabili rappresentano le derivate parziali della
funzione di stato rispetto alle diverse variabili. Questo pu essere chiarito con un semplice
esempio come mostrato nel seguito
64
.
Considerando per esempio la relazione (218) vero che:
n
n
,
,
S
V
V
U
P
S
U
T
|
.
|
\
|
=
|
.
|
\
|
=
(241)
Poich le derivate parziali seconde rispetto a due diverse variabili indipendenti devono
essere uguali indipendentemente dallordine di derivazione, ne consegue che:
S
P
V S
U
S V
U
V
T
2 2
(242)
Applicando la stessa logica ai differenziali esatti di diverse funzioni di stato si possono
ottenere le seguenti relazioni:
n
n
n n
, ,
, ,
, ,
,
,
,
S V S
V V S
V S
V n
P
n V
S n
T
n S dn PdV TdS dU
S
P
V
T
V S
|
.
|
\
|
= |
.
|
\
|
|
.
|
\
|
= |
.
|
\
|
+ =
|
.
|
\
|
= |
.
|
\
|
(243)
63
James Clerk Maxwell, 1831 - 1879, fisico scozzese.
64
Si veda lAppendice A per i dettagli matematici.
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
96
n
n
n n
, ,
, ,
, ,
,
,
,
S P S
P P S
P S
P n
V
n P
S n
T
n S dn VdP TdS dH
S
V
P
T
P S
|
.
|
\
|
= |
.
|
\
|
|
.
|
\
|
= |
.
|
\
|
+ + =
|
.
|
\
|
= |
.
|
\
|
(244)
n
n
n n
, ,
, ,
, ,
,
,
,
T V T
V V T
V T
V n
P
n V
T n
S
n T dn PdV SdT dA
T
P
V
S
V T
|
.
|
\
|
= |
.
|
\
|
|
.
|
\
|
= |
.
|
\
|
+ =
|
.
|
\
|
= |
.
|
\
|
(245)
n
n
n n
, ,
, ,
, ,
,
,
,
T P T
P P T
P T
P n
V
n P
T n
S
n T dn VdP SdT dG
T
V
P
S
P T
|
.
|
\
|
= |
.
|
\
|
|
.
|
\
|
= |
.
|
\
|
+ + =
|
.
|
\
|
= |
.
|
\
|
(246)
Altre relazioni analoghe si possono ricavare dalle espressioni dei differenziali esatti delle
funzioni termodinamiche scritte per sistemi multicomponente.
Come verr mostrato nel seguito del testo, lutilit delle relazioni di Maxwell consiste
essenzialmente nel manipolare algebricamente delle relazioni termodinamiche per ottenere
delle espressioni di una grandezza in funzione di variabili misurabili.
4.2 Principio della minima energia
Si consideri un sistema chiuso, in cui cio il numero di moli costante, che scambi
reversibilmente calore e lavoro con lambiente (assimilabile a un serbatoio di calore e
lavoro
65
). La variazione di entropia dellambiente sar pari a:
65
Analogamente al serbatoio di calore, un serbatoio di lavoro un sistema in grado di scambiare
lavoro senza variare la propria pressione.
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
97
T
dQ
T
dQ
dS
A
A
A
= = (247)
dove lapice
A
indica lambiente e si considerato che la temperatura del sistema e quella
dellambiente devono essere uguali (perch lo scambio di calore sia reversibile) e che il
calore uscente dal sistema pari (a meno del segno) a quello entrante nellambiente.
Poich lunione del sistema e dellambiente un sistema isolato, deve essere:
0 = + =
T
dQ
dS dS dS dS
A tot
(248)
Introducendo nellequazione precedente la forma del primo principio della termodinamica
(che vale per sistemi chiusi) per processi reversibili, equazione (56), si ottiene la seguente
relazione generale:
0 PdV dU TdS (249)
Questa relazione coinvolge solo funzioni di stato e quindi deve essere soddisfatta per ogni
trasformazione in un sistema chiuso. In condizioni di equilibrio vale luguaglianza e si
ritorna alla combinazione del primo e secondo principio della termodinamica per processi
reversibili.
Per un sistema soggetto al vincolo di energia interna e volume costante (cio un sistema
isolato), si ha che dU=dV=0 e la relazione precedente diventa:
0
,
V U
dS
(250)
che implica che lentropia pu solo aumentare per trasformazioni spontanee e quindi, in
condizioni di equilibrio, deve essere massima.
Per un sistema soggetto invece al vincolo di entropia e volume costante, si ha che dS=dV=0
e la relazione (249) diventa:
0
,
V S
dU
(251)
che implica che lenergia interna pu solo diminuire e quindi, in condizioni di equilibrio,
deve essere minima.
Per un sistema soggetto al vincolo di temperatura e volume costante, si ha che dT=dV=0 e
la relazione (249) diventa:
( ) ( ) 0 = = = dA U TS d dU TS d PdV dU TdS (252)
cio:
0
,
V T
dA
(253)
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
98
che implica che lenergia libera di Helmholtz pu solo diminuire e quindi, in condizioni di
equilibrio, deve essere minima.
Infine, per un sistema soggetto al vincolo di temperatura e pressione costante, si ha che
dT=dP=0 e la relazione (249) diventa:
( ) ( ) ( ) 0 = = = dG PV U TS d PV d dU TS d PdV dU TdS (254)
cio:
0
,
P T
dG
(255)
che implica che lenergia libera di Gibbs pu solo diminuire e quindi, in condizioni di
equilibrio, deve essere minima.
Relazioni analoghe valgono ovviamente anche per le grandezze specifiche. Riassumendo,
le condizioni di equilibrio per un sistema chiuso soggetto a diversi vincoli sono le seguenti:
( )
( )
( )
( ) 0 ; 0 : min cost ,
0 ; 0 : min cost ,
0 ; 0 : min cost ,
0 ; 0 : max cost ,
2
2
2
2
> = =
> = =
> = =
< = =
g d dg g P T
a d da a v T
u d du u v s
s d ds s v u
(256)
Analoghe relazioni valgono ovviamente per le grandezze estensive. Mentre, nello stato di
equilibrio, lentropia deve assumere il valore massimo compatibile coi vincoli, le diverse
energie devono assumere il valore minimo: questo rappresenta il principio della minima
energia.
4.3 Funzioni residue
La relazione fondamentale per un fluido puro omogeneo in un sistema chiuso la
equazione (240):
vdP sdT dg + =
(257)
Questa relazione consente il calcolo della variazione dellenergia libera di Gibbs molare
sulla base della variazione di temperatura e pressione.
Risulta comodo utilizzare la forma adimensionale dellenergia libera di Gibbs molare
( ) RT g . Differenziando questa funzione adimensionale si ottiene:
dT
RT
g
dg
RT RT
g
d
2
1
= |
.
|
\
|
(258)
Inserendo in questa identit la relazione (257) e la definizione di g=h-Ts, si ottiene:
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
99
dT
RT
h
dP
RT
v
RT
g
d
2
= |
.
|
\
|
(259)
Questa relazione, che ha il vantaggio di contenere solo termini adimensionali, consente il
calcolo di tutte le funzioni termodinamiche specifiche (sempre in forma adimensionale)
dalla conoscenza della sola energia libera di Gibbs specifica, g(T,P):
( )
T
P
RT g
RT
v
(
=
(260)
( )
P
T
RT g
T
RT
h
(
=
(261)
RT
g
RT
h
R
s
= (262)
RT
Pv
RT
h
RT
u
= (263)
In altri termini, g pu essere vista come la funzione generatrice di tutte le altre grandezze
termodinamiche poich implicitamente contiene tutte le informazioni sullo stato intensivo
del sistema.
Il problema ovviamente che g non una grandezza misurabile e deve quindi essere
calcolata dalle grandezze misurabili sulla base di relazioni termodinamiche esatte. Si noti
che lequazione (260) implica lesistenza di una relazione tra g e v; in altri termini, noto v
(una grandezza misurabile) deve essere possibile poter calcolare g. Pi usualmente, poich
possibile calcolare g per un gas perfetto utilizzando le relazioni dedotte per il calcolo di h
e s (equazioni (107) e (109)) come:
( ) ( ) ( )
( ) ( )
|
|
|
.
|
\
|
|
|
.
|
\
|
+ + =
= =
r
T
T
P
r r
T
T
P r
P
P
R dT
T
c
P T s T dT c T h
P T Ts T h P T g
r r
ln ,
, ,
*
* * *
* * *
(264)
(dove col pedice
r
si indicato lo stato di riferimento) si preferisce calcolare lo scostamento
di g dal valore che avrebbe un gas perfetto nelle stesse condizioni. Tale scostamento prende
il nome di funzione residua, che viene quindi definita per una generica grandezza m come:
( ) ( ) ( ) P T m P T m P T m
R
, , ,
*
= (265)
Risulta evidente che, poich i valori delle funzioni termodinamiche di un gas perfetto sono
facilmente calcolabili (una volta che sia nota la funzione ( ) T c
P
*
, cio il valore del calore
specifico a pressione costante in funzione della temperatura), dal valore della funzione
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
100
residua si pu facilmente risalire al valore della funzione. Il differenziale della funzione
g
R
/RT viene dedotto dalla relazione (259):
dT
RT
h
dP
RT
v
dT
RT
h
dP
RT
v
dT
RT
h
dP
RT
v
RT
g
d
RT
g
d
RT
g
d
R R
R
2 2
* *
2
*
=
|
|
.
|
\
|
=
=
|
|
.
|
\
|
|
.
|
\
|
=
|
|
.
|
\
|
(266)
Noto il valore residuo della funzione g/RT possono essere calcolati anche i valori residui
delle altre funzioni termodinamiche con relazioni analoghe a quelle viste in precedenza:
( )
T
R R
P
RT g
RT
v
(
(
=
(267)
( )
P
R R
T
RT g
T
RT
h
(
(
=
(268)
RT
g
RT
h
R
s
R R R
=
(269)
RT
Pv
RT
h
RT
u
R R R
=
(270)
Come al solito, il valore di una funzione di stato viene calcolato seguendo il percorso pi
comodo. In questo caso, il valore della funzione g/RT pu essere calcolato considerando
una trasformazione isoterma:
cost
1
0
) (
0
) (
0
=
|
|
.
|
\
|
= =
|
|
.
|
\
|
T dP
RT
v
dg
RT RT
g
d
P
R
P g
R
P g
R
R R
(271)
Lestremo inferiore di integrazione nasce dal fatto che per pressioni prossime a zero tutti i
fluidi si comportano come un gas perfetto, e quindi le grandezze residue diventano per
definizione nulle.
La relazione precedente fornisce la seguente:
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
101
( )
|
.
|
\
|
=
= |
.
|
\
|
=
|
|
.
|
\
|
=
P
P P
R
P
dP
RT
Pv
dP
RT
P RT
RT
v
dP
RT
v v
RT
P T g
0
0 0
*
1
/
0
,
(272)
Introducendo il coefficiente di compressibilit la relazione precedente diventa:
( )
( ) cost 1
,
0
= =
T
P
dP
Z
RT
P T g
P
R
(273)
Limportante implicazione di questa equazione che sulla base della conoscenza di
unequazione di stato (cio della funzione Z(T,P), sviluppata da misure sperimentali P v
T) possibile calcolare il valore dellenergia libera di Gibbs residua e quindi di tutte le altre
funzioni residue. In altri termini, dallequazione di stato possibile calcolare tutte le
funzioni termodinamiche dei fluidi puri. Questo spiega lenorme importanza delle
equazioni di stato nella termodinamica dellingegneria chimica.
Ovviamente, per un gas perfetto Z=1 e g
R
=0.
Utilizzando le relazioni (268) e (269) insieme alla (273) si possono ricavare le seguenti
espressioni per il calcolo dellentalpia e dellentropia residua specifica:
( )
cost
0
= |
.
|
\
|
=
(
(
=
T
P
dP
T
Z
T
T
RT g
T
RT
h
P
P
P
R R
(274)
( ) cost 1
0 0
= |
.
|
\
|
= =
T
P
dP
Z
P
dP
T
Z
T
RT
g
RT
h
R
s
P P
P
R R R
(275)
Anche in questo caso, nota unequazione di stato, cio una relazione funzionale Z=Z(T,P)
per il fluido in esame, possibile calcolare lentalpia e lentropia residua specifica a
qualsiasi temperatura e pressione. Dallespressione delle grandezze residue si risale poi
facilmente al valore della grandezza:
( ) ( ) ( ) ( ) ( ) P T h dT c T h P T h T h P T h
R
T
T
P r
R
r
, , ,
* * *
+ + = + =
(276)
( ) ( ) ( ) ( ) ( ) P T s
P
P
R dT
T
c
P T s P T s P T s P T s
R
r
T
T
P
r r
R
r
, ln , , , ,
*
* *
+
|
|
.
|
\
|
+ = + =
(277)
La variazione di entalpia ed entropia di un fluido reale tra due stati (T
1
, P
1
) e (T
2
, P
2
) si pu
quindi calcolare come:
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
102
( ) ( ) ( ) ( )
( ) ( )
1 1 2 2
*
1 1
* *
2 2
* *
1 1 2 2 2 1
, ,
, ,
) , ( ) , (
2
1
1 2
P T h P T h dT c
P T h dT c T h P T h dT c T h
P T h P T h h
R R
T
T
P
R
T
T
P r
R
T
T
P r
r r
+ =
=
(
(
+ +
(
(
+ + =
= =
(278)
( ) ( )
( ) ( )
( ) ( )
1 1 2 2
1
2
*
1 1
1
*
*
2 2
2
*
*
1 1 2 2 2 1
, , ln
, ln ,
, ln ,
) , ( ) , (
2
1
1
2
P T s P T s
P
P
R dT
T
c
P T s
P
P
R dT
T
c
P T s
P T s
P
P
R dT
T
c
P T s
P T s P T s s
R R
T
T
P
R
r
T
T
P
r r
R
r
T
T
P
r r
r
r
+
|
|
.
|
\
|
=
=
(
(
+
|
|
.
|
\
|
+
+
(
(
+
|
|
.
|
\
|
+ =
= =
(279)
Gli integrali presenti nelle relazioni (274) e (275) possono essere calcolati utilizzando le
equazioni di stato presentate nel capitolo precedente, come discusso nelle sezioni seguenti.
In conclusione, dalla conoscenza del calore specifico in funzione della temperatura e di una
equazione di stato possibile calcolare la variazione di tutte le funzioni termodinamiche di
un fluido reale tra due stati. Il fluido si pu trovare in diverse fasi nei due stati considerati:
solida, liquida o vapore. Alcune delle equazioni di stato discusse nel capitolo precedente
sono in grado di rappresentare il comportamento volumetrico sia della fase vapore sia di
quella liquida; di conseguenza, le funzioni residue consentono in calcolo delle grandezze
termodinamiche sia della fase vapore, sia di quella liquida. A pressioni maggiori della
tensione di vapore alla temperatura considerata si otterr il valore delle funzioni residue
relative alla fase liquida, a pressioni inferiori quello della fase vapore. Alla pressione pari
alla tensione di vapore si otterranno due valori delle funzioni residue: quello del liquido
saturo e quello del vapore saturo, rispettivamente.
Per il calcolo delle grandezze termodinamiche delle fasi condensate anche possibile un
approccio alternativo, basato sulla conoscenza dei calori latenti di transizione di fase.
Questo verr illustrato nella sezione 4.5.
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
103
4.3.1 EoS del viriale
Per pressioni non troppo elevate e per la fase gassosa lequazione di stato del viriale
fornisce delle previsioni ragionevoli del coefficiente di compressibilit con la forma
troncata al secondo coefficiente
66
:
RT
BP
Z + 1 (280)
da cui si ricava che:
dT
dB
RT
P
RT
BP
T
Z
P
+ = |
.
|
\
|
2
(281)
Le funzioni residue si calcolano quindi come:
( )
RT
BP
P
dP
RT
BP
P
dP
Z
RT
g
P P
R
= |
.
|
\
|
+ = =
0 0
1 1 1 (282)
( ) ( )
|
.
|
\
|
=
= |
.
|
\
|
=
(
=
(
(
=
dT
dB
T B
RT
P
T
B
dT
dB
T R
TP
T
RT BP
T
T
RT g
T
RT
h
P
P
R R
2
1
(283)
dT
dB
R
P
RT
BP
dT
dB
T B
RT
P
RT
g
RT
h
R
s
R R R
= |
.
|
\
|
= = (284)
I valori del secondo coefficiente viriale e della sua derivata rispetto alla temperatura si
possono calcolare con le correlazioni (175) e (176):
( ) ( )
( )
( )
2 , 4
1
6 , 1
0
1 0
172 , 0
139 , 0
422 , 0
083 , 0
R
R
T
T B
T
T B
T B T B
RT
BP
R
R
R R
C
C
=
=
+ =
(285)
da cui:
66
Si ricordi che il secondo coefficiente del viriale B dipende solo dalla temperatura.
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
104
|
|
|
.
|
\
|
+ =
|
|
.
|
\
|
+ =
=
|
|
.
|
\
|
=
|
|
.
|
\
|
=
|
|
.
|
\
|
=
2 , 5 6 , 2
1 0
722 , 0 675 , 0
1
R R
T T
P
R
dT
dB
dT
dB
P
R
T RT
BP
dT
d
P
RT
dT
dT
RT
BP
dT
d
P
RT
RT
BP
dT
d
P
RT
dT
dB
C R R C
C C
C
R C
C R
C
C
R C
C
C
C
C
C
(286)
Note le variabili critiche quindi possibile calcolare le funzioni residue per qualsiasi fluido,
almeno nella regione in cui sono valide le relazioni (285).
4.3.1.1 Coefficiente di Joule - Thomson
67
Una chiara evidenza del comportamento di un gas diverso da quello del gas perfetto lo si ha
dal classico esperimento di Joule e Thomson in cui un gas viene fatto espandere attraverso
un setto poroso o una valvola posta in un condotto ben coibentato misurando la temperatura
e la pressione a monte e a valle.
Come discusso nella sezione 2.3.1, lespansione di un gas attraverso una valvola un
processo isoentalpico, cio lentalpia specifica della corrente a monte pari a quella della
corrente a valle della valvola.
Poich per un gas perfetto lentalpia dipende solo dalla temperatura, anche la temperatura
delle due correnti deve essere uguale. In altri termini, lespansione isoentalpica di un gas
perfetto non ne cambia la temperatura.
Sperimentalmente si nota invece che la temperatura di un gas pu sia aumentare sia
diminuire al variare della pressione in condizioni isoentalpiche
68
; la variazione di
temperatura che un fluido presenta al variare della pressione in condizioni isoentalpiche
viene chiamato coefficiente di Joule - Thomson:
h
P
T
|
.
|
\
|
= (287)
Il suo valore pu essere positivo (il fluido si raffredda espandendosi) oppure negativo (il
fluido si raffredda comprimendosi), in funzione del tipo di fluido e delle condizioni di
temperatura e pressione. Il coefficiente di Joule - Thomson pu essere calcolato utilizzando
unequazione di stato come segue.
Poich lo stato intensivo di un sistema omogeneo monocomponente definito una volta che
sono assegnate due variabili intensive, lentalpia specifica deve essere funzione di due sole
67
Lord William Thomson Kelvin, 1824 - 1907, matematico e fisico irlandese.
68
Questo fenomeno pu essere sfruttato per liquefare un gas.
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
105
variabili intensive, per esempio temperatura e pressione: h = h(T,P). Essendo lentalpia una
funzione di stato il suo differenziale esatto; in altri termini, la variazione infinitesima di
entalpia legata alla variazione infinitesima di temperatura e pressione attraverso le
relazione formale:
dP
P
h
dT c dP
P
h
dT
T
h
dh
T
P
T P
|
.
|
\
|
+ = |
.
|
\
|
+ |
.
|
\
|
=
(288)
dove si utilizzata la definizione di calore specifico a pressione costante, equazione (64). In
condizioni isoentalpiche dh = 0 e la relazione precedente diventa:
P
h
P
T
c
P
T
c
P
h
= |
.
|
\
|
= |
.
|
\
|
(289)
Dalla relazione (229) scritta in termini specifici e derivata rispetto alla pressione a
temperatura costante si ricava:
v
T
v
T v
P
s
T
P
h
vdP Tds dh
P T T
+ |
.
|
\
|
= + |
.
|
\
|
= |
.
|
\
|
=
(290)
Lultimo passaggio possibile grazie alla relazione di Maxwell (246) scritta in termini
specifici. Inserendo questa equazione nella relazione (290) si ottiene:
|
|
.
|
\
|
|
.
|
\
|
= |
.
|
\
|
= v
T
v
T
c P
h
c
P P T P
1 1
(291)
Nel caso di gas perfetto si ottiene ovviamente
0
1
= |
.
|
\
|
=
P
RT
P
R
T
c
P
(292)
La relazione (291) pu essere espressa in funzione del coefficiente di compressibilit:
P P P P
T
Z
Pc
RT
P
ZRT
P
ZR
T
Z
P
RT
T
c
|
.
|
\
|
=
|
|
.
|
\
|
|
|
.
|
\
|
+ |
.
|
\
|
=
2
1
(293)
che, inserendo la relazione (281), diventa:
|
.
|
\
|
= |
.
|
\
|
+ = B
dT
dB
T
c dT
dB
RT
P
RT
BP
Pc
RT
P P
1
2
2
(294)
Utilizzando le relazioni (285) e (286) possibile calcolare il coefficiente di Joule -
Thomson di un qualsiasi fluido e prevederne quindi il comportamento termico a seguito di
unespansione isoentalpica.
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
106
4.3.2 EoS cubiche
Utilizzando lEoS del viriale possibile ricavare delle espressioni esplicite delle funzioni
residue in quanto tale EoS fornisce in modo esplicito la funzione Z=Z(P) con lequazione
(280), consentendo cos il calcolo analitico della derivata rispetto alla temperatura e
dellintegrale nella pressione presenti nelle equazioni (274) e (275).
Purtroppo le EoS cubiche sono esplicite solo nella pressione e non consentono di ricavare
in modo esplicito la relazione Z=Z(P). La relazione (273) non quindi utilizzabile
direttamente con equazioni di stato cubiche.
Risulta perci necessario manipolare tale relazione per trasformarla in una forma
utilizzabile con le EoS cubiche. Questo pu essere fatto utilizzando un cambiamento di
variabili basato sulla relazione che definisce il coefficiente di compressibilit differenziata a
temperatura costante:
RTdZ vdP Pdv ZRT Pv = + = (295)
Dividendo membro a membro questa relazione per Pv (a sinistra) e ZRT (a destra) si
ottiene:
v
dv
Z
dZ
P
dP
Z
dZ
P
dP
v
dv
= = + (296)
Sostituendo questa espressione nella relazione (273) e tenendo conto che 1 ) 0 ( = = P Z e
= = ) 0 (P v si ottiene:
( ) ( ) ( )
( )
=
= = =
v
v Z P
R
v
dv
Z Z Z
v
dv
Z
Z
dZ
Z
P
dP
Z
RT
g
1 ln 1
1 1 1
1 0
(297)
Analogamente, tralasciando i dettagli matematici
69
, le relazioni precedenti possono essere
poste nella seguente forma equivalente:
cost
1
1 =
|
|
.
|
\
|
|
.
|
\
|
+ =
T dv P
T
P
T
RT
Z
RT
h
v
v
R
(298)
69
Si veda per tali dettagli per esempio il testo H.C. van Ness e M.M. Abbott Classical
thermodynamics of nonelectrolyte solutions, McGraw Hill, 1982.
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
107
cost
1 1
ln =
|
|
.
|
\
|
|
.
|
\
|
+ =
T dv
v T
P
R
Z
R
s
v
v
R
(299)
Queste relazioni possono essere utilizzate con le diverse EoS cubiche per ottenere delle
relazioni esplicite delle funzioni residue. Per esempio, utilizzando lEoS di van der Waals
(equazione (478)) si ha che:
b v
R
T
P
v
a
b v
RT
P
v
= |
.
|
\
|
=
2
(300)
e quindi
Z
A
Z
RTv
a
Z dv
v
a
RT
Z
dv
v
a
b v
RT
b v
R
T
RT
Z
RT
h
v
v
R
= = + =
=
|
|
.
|
\
|
|
.
|
\
|
+ =
1 1
1
1
1
1
2
2
(301)
Il principale vantaggio delle EoS cubiche quello di prevedere il comportamento
volumetrico sia della fase vapore, sia di quella liquida. Di conseguenza, possibile
calcolare le funzioni residue di entrambe le fasi. A titolo di esempio, la procedura per il
calcolo dellentalpia residua della fase liquida o vapore con lequazione precedente la
seguente:
per un dato fluido si calcolano, tramite i valori della temperatura e della pressione
critica, i parametri a e b dellEoS di van der Waals;
assegnati i valori di temperatura e pressione si calcolano i relativi valori dei parametri
A e B e si risolve lEoS cubica in Z; se si trova un solo valore reale positivo si utilizza
tale valore nellequazione (301) e si calcola lentalpia residua; se viceversa si trovano
tre valori reali positivi si utilizza il minore se si vuole calcolare lentalpia residua del
liquido e il maggiore se si vuole calcolare quella del vapore;
in entrambi i casi (liquido o vapore) si somma poi il valore dellentalpia residua a
quella del gas perfetto alla stessa temperatura e pressione per calcolare il valore
dellentalpia.
Il calcolo delle funzioni residue utilizzando altre EoS cubiche procede in modo simile sulla
base di relazioni differenti dedotte con procedimenti analoghi a quello illustrato per
lentalpia residua con la EoS di van der Waals. Tali relazioni sono riassunte in Tabella 8,
Tabella 9 e Tabella 10.
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
108
EoS h
R
/(RT)
vdW
Z
A
Z 1
RK |
.
|
\
| +
Z
B Z
B
A
Z ln
2
3
1
RKS ( ) |
.
|
\
| +
+
Z
B Z
B
A
Z ln 1 1
PR ( )
( )
( )
|
|
.
|
\
|
+
+ +
+
2 1
2 1
ln 1
2 2
1
B Z
B Z
B
A
Z
Tabella 8: espressioni per il calcolo dellentalpia residua ( k T S
R
= ).
EoS s
R
/R
vdW ( ) B Z ln
RK ( ) |
.
|
\
| +
Z
B Z
B
A
B Z ln
2
ln
RKS ( ) |
.
|
\
| +
Z
B Z
B
A
B Z ln ln
PR ( )
( )
( )
|
|
.
|
\
|
+
+ +
2 1
2 1
ln
2 2
ln
B Z
B Z
B
A
B Z
Tabella 9: espressioni per il calcolo dellentropia residua ( k T S
R
= ).
EoS g
R
/(RT)
vdW ( ) B Z
Z
A
Z ln 1
RK ( ) B Z
Z
B Z
B
A
Z |
.
|
\
| +
ln ln 1
RKS ( ) B Z
Z
B Z
B
A
Z |
.
|
\
| +
ln ln 1
PR
( )
( )
( ) B Z
B Z
B Z
B
A
Z
|
|
.
|
\
|
+
+ +
ln
2 1
2 1
ln
2 2
1
Tabella 10: espressioni per il calcolo dellenergia libera di Gibbs residua ( k T S
R
= ).
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
109
4.3.3 Equazione degli stati corrispondenti
Le relazioni generali per il calcolo delle funzioni residue (274) e (275) possono essere poste
in una forma generalizzata inserendo le definizioni delle variabili ridotte:
R
P
R C P
R C
R C
R C
R C
T
Z
T T
Z
dT T dT
dP P dP
T T T
P P P
|
|
.
|
\
|
= |
.
|
\
|
=
=
=
=
1
(302)
Le relazioni risultanti dopo aver semplificato e raccolto a destra delluguale tutti i termini
che dipendono solo dalle variabili ridotte sono le seguenti:
|
|
.
|
\
|
=
R
R
R
P
R
R
P
R C
R
P
dP
T
Z
T
RT
h
0
2
(303)
( )
|
|
.
|
\
|
=
R R
R
P
R
R
P
R
R
P
R
R
R
P
dP
Z
P
dP
T
Z
T
R
s
0 0
1 (304)
Si nota che vi sono due gruppi adimensionali,
C
R
RT h e R s
R
, che dipendono solo dalla
temperatura e pressione ridotta, oltre che dal coefficiente di compressibilit, Z, e dalle sue
derivate.
La legge degli stati corrispondenti a tre parametri (171)
( ) ( )
R R R R
P T Z P T Z Z , ,
1 0
+ = (305)
fornisce una espressione della derivata di Z:
R R R P
R
P
R
P
R
T
Z
T
Z
T
Z
|
|
.
|
\
|
+
|
|
.
|
\
|
=
|
|
.
|
\
|
1 0
(306)
che inserita nelle equazioni precedenti porta alle seguenti relazioni:
|
|
.
|
\
|
|
|
.
|
\
|
=
R
R
R
R
R
R
P
R
R
P
R
P
R
R
P
R C
R
P
dP
T
Z
T
P
dP
T
Z
T
RT
h
0
1
2
0
0
2
(307)
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
110
|
|
|
.
|
\
|
+
|
|
.
|
\
|
|
|
|
.
|
\
|
+
|
|
.
|
\
|
=
R
R
R
R
P
R
R
P
R
R
P
R
R
P
R
R
R
P
dP
Z
T
Z
T
P
dP
Z
T
Z
T
R
s
0
1
1
0
0
0
1 1 (308)
Il primo integrale del termine a destra di queste due equazioni pu essere valutato in
funzione delle sole variabili T
R
e P
R
dai dati tabulati di Z
0
(T
R
,P
R
). Analogamente, il secondo
integrale pu essere valutato in funzione delle sole variabili T
R
e P
R
dai dati tabulati di
Z
1
(T
R
,P
R
). Questo significa che le due relazioni precedenti contengono dei termini che sono
funzione solo della temperatura e pressione ridotta, usualmente definiti come:
|
|
.
|
\
|
=
(
(
|
|
.
|
\
|
=
(
(
R
R
R
R
R
R
P
R
R
P
R C
R
P
R
R
P
R C
R
P
dP
T
Z
T
RT
h
P
dP
T
Z
T
RT
h
0
1
2
1
0
0
2
0
(309)
|
|
|
.
|
\
|
+
|
|
.
|
\
|
=
(
(
|
|
|
.
|
\
|
+
|
|
.
|
\
|
=
(
(
R
R
R
R
P
R
R
P
R
R
R
P
R
R
P
R
R
R
P
dP
Z
T
Z
T
R
s
P
dP
Z
T
Z
T
R
s
0
1
1
1
0
0
0
0
1
1
(310)
Queste quattro funzioni dipendono quindi solo dalle variabili ridotte e possono essere
riportati in grafici o tabelle analogamente a quanto fatto per le due funzioni Z
0
(T
R
,P
R
) e
Z
1
(T
R
,P
R
). I valori proposti per queste quattro funzioni da Lee e Kesler sono riportati in
Appendice C.
Analogamente al coefficiente di compressibilit quindi possibile calcolare le funzioni
residue dai valori delle variabili ridotte e del fattore acentrico di Pitzer:
1 0
(
(
+
(
(
=
C
R
C
R
C
R
RT
h
RT
h
RT
h
(311)
1 0
(
(
+
(
(
=
R
s
R
s
R
s
R R R
(312)
Analogamente al caso del coefficiente di compressibilit, le funzioni | |
1
C
R
RT h e | |
1
R s
R
rappresentano una correzione ai valori delle funzioni | |
0
C
R
RT h e | |
0
R s
R
e possono
quindi essere spesso trascurate per una valutazione di massima.
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
111
4.3.4 Applicazione
Si calcolino i valori residui dellentalpia, dellentropia e dellenergia libera di Gibbs del n-
ottano in fase vapore a 427.9 [K] e 2.15 [bar] con le diverse equazioni di stato viste in
questo capitolo.
Si noti che nellApplicazione 3.4 si sono gi calcolati i valori di Z, A, B, S, k, T
R
e P
R
che
verranno riutilizzati in questa applicazione.
Gas ideale
Per definizione di funzione residua:
0 = = =
R R R
g s h (313)
vdW
0643 , 0 = A
0143 , 0 = B
(314)
0,9475 = Z (315)
1204 , 0
9475 , 0
0643 , 0
1 9475 , 0 1 = = =
Z
A
Z
RT
h
R
(316)
( ) ( ) 0692 , 0 0143 , 0 9475 , 0 ln ln = = = B Z
R
s
R
(317)
] mol [J 43 , 428 9 , 427 314 , 8 0692 , 0
1
= = = RT
RT
h
h
R
R
(318)
] K mol [J 58 , 0 314 , 8 0692 , 0
1 1
= = = R
R
s
s
R
R
(319)
( ) ] mol [J 24 , 182 58 , 0 9 , 427 43 , 428
1
= = =
R R R
Ts h g (320)
RK
0752 , 0 = A
0099 , 0 = B
(321)
0,9309 = Z (322)
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
112
1897 , 0 ln
2
3
1 = |
.
|
\
| +
=
Z
B Z
B
A
Z
RT
h
R
(323)
( ) 1226 , 0 ln
2
ln = |
.
|
\
| +
=
Z
B Z
B
A
B Z
R
s
R
(324)
] mol [J 68 , 674
1
= = RT
RT
h
h
R
R
(325)
] K mol [J 02 , 1
1 1
= = R
R
s
s
R
R
(326)
] mol [J 76 , 238
1
= =
R R R
Ts h g (327)
RKS
0786 , 1 = S
3078 , 1 = k
(328)
( )
0852 , 0
2
= =
RT
aP
A
0099 , 0 = =
RT
bP
B
(329)
0,9192 = Z (330)
2485 , 0 ln 1 1 = |
.
|
\
| +
|
|
.
|
\
|
+ =
Z
B Z
k
T
S
B
A
Z
RT
h
R
R
(331)
( ) 1705 , 0 ln ln = |
.
|
\
| +
=
Z
B Z
B
k T AS
B Z
R
s
R
R
(332)
] mol [J 91 , 883
1
= = RT
RT
h
h
R
R
(333)
] K mol [J 42 , 1
1 1
= = R
R
s
s
R
R
(334)
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
113
] mol [J 20 , 277
1
= =
R R R
Ts h g (335)
PR
9457 , 0 = S
2676 , 1 = k
(336)
0837 , 0 = A
0089 , 0 = B
(337)
0,9151 = Z (338)
( )
( )
2502 , 0
2 1
2 1
ln 1
2 2
1 =
|
|
.
|
\
|
+
+ +
|
|
.
|
\
|
+ =
B Z
B Z
k
T
S
B
A
Z
RT
h
R
R
(339)
( )
( )
( )
1682 , 0
2 1
2 1
ln
2 2
ln =
|
|
.
|
\
|
+
+ +
=
B Z
B Z
B
k T AS
B Z
R
s R
R
(340)
] mol [J 02 , 890
1
= = RT
RT
h
h
R
R
(341)
] K mol [J 40 , 1
1 1
= = R
R
s
s
R
R
(342)
] mol [J 83 , 291
1
= =
R R R
Ts h g (343)
Stati corrispondenti
Dalle tabelle di Lee - Kesler a T
R
= 0,75 e P
R
= 0,086 si ricava (interpolando i valori
riportati):
16 , 0
0
=
(
(
C
R
RT
h
26 , 0
0
=
(
(
C
R
RT
h
(344)
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
114
( ) 26 , 0 26 , 0 398 , 0 16 , 0
1 0
= + =
(
(
+
(
(
=
C
R
C
R
C
R
RT
h
RT
h
RT
h
(345)
16 , 0
0
=
(
(
C
R
RT
h
29 , 0
1
=
(
(
R
s
R
(346)
( ) 26 , 0 29 , 0 398 , 0 14 , 0
1 0
= + =
(
(
+
(
(
=
R
s
R
s
R
s
R R R
(347)
] mol [J 00 , 1232 569,4 314 , 8 26 , 0
1
= = =
C
C
R
R
RT
RT
h
h (348)
] K mol [J 12 , 2 314 , 8 26 , 0
1 1
= = = R
R
s
s
R
R
(349)
] mol [J 58 , 324
1
= =
R R R
Ts h g (350)
Viriale
7555 , 0 =
C
C
RT
BP
6797 , 2
722 , 0 675 , 0
2 , 5 6 , 2
=
|
|
|
.
|
\
|
+ =
C C
P
R
T T
P
R
dT
dB
R R
(351)
3180 , 0 6797 , 2 = = |
.
|
\
|
=
R
C
C
R
R
R
P
RT
BP
T
P
dT
dB
T B
RT
P
RT
h
(352)
2315 , 0 6797 , 2 = = =
R
R
P
dT
dB
R
P
R
s
(353)
] mol [J 1131,40
1
= = RT
RT
h
h
R
R
(354)
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
115
] K mol [J 92 , 1
1 1
= = R
R
s
s
R
R
(355)
] mol [J 95 , 307
1
= =
R R R
Ts h g (356)
I risultati ottenuti sono riassunti nella Tabella 11. Si nota che non vi sono grandi differenze
tra le diverse equazioni di stato in quanto il comportamento del fluido prossimo a quello
di un gas perfetto.
Seguendo lo stesso procedimento anche possibile calcolare le grandezze residue per il n-
ottano liquido alla stessa temperatura ma a una pressione di 5 [bar]. Questa volta, come
riportato in Tabella 11, le differenze sono marcate. In particolare, le equazioni di stato del
gas perfetto e del viriale forniscono ovviamente risultati non corretti. Inoltre, anche le
equazioni di van der Waals e RK forniscono risultati diversi da quelli delle altre equazioni,
che risultano viceversa abbastanza equivalenti.
Lo svolgimento di questo tipo di conti ancora una volta tedioso, soprattutto se
necessario ripeterli un gran numero di volte. Analogamente a quanto fatto per il calcolo del
volume molare, risulta utile implementare la sequenza dei calcoli in un programma di
calcolo. A titolo di esempio, si riporta di seguito un programma in linguaggio MATLAB, che
deriva da un ampliamento del programma vTP discusso nellApplicazione 3.4.
Gas, 427,9 [K] e 2,15 [bar] Liquido, 427,9 [K] e 5 [bar]
EoS h
R
s
R
g
R
h
R
s
R
g
R
Gas perfetto 0,00 0,00 0,00 0,00 0,00 0,00
vdW -428,4 -0,57 -182,2 -14179 -34,39 536,3
RK -674,7 -1,02 -238,8 -24316 -53,73 -1327,0
RKS -883,9 -1,42 -277,2 -33729 -71,24 -3243,2
PR -890,0 -1,40 -291,8 -33363 -70,28 -3289,6
Stati corrispondenti -1232,0 -2,12 -324,6 -33109 -69,61 -3321,0
Viriale -1131,4 -1,92 -308,0 -2631 -4,47 -716,2
Tabella 11: valori delle funzioni residue in [J mol
-1
] e [J mol
-1
K
-1
] calcolati con le diverse
EoS.
Il programma commentato e quindi di facile comprensione e pu essere implementato in
un file testo di nome vhsgTP.m per essere eseguito in ambiente MATLAB. Il programma
EoS richiamato lo stesso utilizzato nellApplicazione 3.4, mentre il calcolo delle funzioni
residue in forma adimensionale demandato alla routine esterna fr, il cui listato riportato
di seguito alla routine vhsgTP e pu essere implementato in un file testo di nome fr.m.
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
116
%vhsgTP calcolo del volume molare
% e delle funzioni residue
% a T e P con diverse EoS
% R. Rota (2002)
format short g %formato scrittura
format compact %formato scrittura
clear all %cancella tutte le variabili
%dati per n-ottano
Tc =569.4; %[K]
Pc =24.97e5; %[Pa]
om =0.398; %fattore acentrico di Pitzer
%calcolo volume molare gas (fase=1) o liquido (fase=2)
fase = 1;
%assegno le condizioni di T e P
T = 427.9; %temperatura [K]
P = 2.15e5; %pressione [Pa]
R = 8.314; %cost. gas [J/(mol K)]
RT =R*T;
RTc=R*Tc;
TR =T/Tc;
PR =P/Pc;
%1 gas perfetto
disp('gas perfetto')
v = R*T/P
hr=0
sr=0
gr=0
pause
%2 vdW, RK, RKS, PR
eq=['vdW';'RK ';'RKS';'PR ']; %nomi EoS
for tipo=1:4 %ciclo su diverse EoS
[Z,A,B,S,k] = EoS(T,P,Tc,Pc,om,tipo); %calcolo Z a (P,T)
STrk = S*sqrt(TR/k);
disp(eq(tipo,:)) %visualizzo nome EoS
if fase == 1
v = Z(3)*RT/P %calcolo Vgas [m3/mol]
[hrRT,srR] = fr(Z(3),A,B,STrk,tipo); %calc. hr/RT e sr/R
hr = hrRT*RT
sr = srR*R
gr = hr - T*sr
else
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
117
v = Z(1)*RT/P %calcolo Vliq [m3/mol]
[hrRT,srR] = fr(Z(1),A,B,STrk,tipo); %calc. hr/RT e sr/R
hr = hrRT*RT
sr = srR*R
gr = hr - T*sr
end
pause
end
%3 stati corrispondenti @ TR=0.75
clear Z; %cancello il vettore Z
%vettori coi valori di Z0 e Z1 a TR=0,75 per varie PR
PRZ=[.01 .05 .1 .2 .4 .6 .8 1 1.2 1.5 2 3 5 7 10];
Z0 =[.9922 .9598 .9165 .0336 .067 .1001 .133 .1656 ...
.1981 .2426 .3260 .4823 .7854 1.0787 1.5047];
Z1 =[-.0064 -.0339 -.0744 -.0143 -.0282 -.0417 -.055 ...
-.0681 -.0808 -.0996 -.1298 -.1872 -.2929 ...
-.3901 -.525];
hrRTC0=[ .017 .088 .183 4.687 4.679 4.672 4.664 ...
4.665 4.646 4.632 4.607 4.554 4.434 4.393 ...
4.095]*(-1);
hrRTC1=[ .027 .142 .306 5.796 5.802 5.809 5.816 ...
5.824 5.832 5.845 5.868 5.918 6.027 6.142 ...
6.318]*(-1);
srR0=[ .015 .078 .164 5.917 5.248 4.866 4.600 ...
4.399 4.238 4.045 3.807 3.491 3.138 2.939 ...
2.761]*(-1);
srR1=[ .029 .156 .340 6.173 6.167 6.162 6.158 ...
6.155 6.152 6.149 6.147 6.149 6.174 6.213 ...
6.285]*(-1);
Z0i=interp1(PRZ,Z0,PR); %interpolo al valore di PR
Z1i=interp1(PRZ,Z1,PR); %interpolo al valore di PR
Z = Z0i + om*Z1i; %calcolo Z
hrRTC0i=interp1(PRZ,hrRTC0,PR); %interpolo al valore di PR
hrRTC1i=interp1(PRZ,hrRTC1,PR); %interpolo al valore di PR
hrRTC = hrRTC0i + om*hrRTC1i; %calcolo hrRTC
srR0i=interp1(PRZ,srR0,PR); %interpolo al valore di PR
srR1i=interp1(PRZ,srR1,PR); %interpolo al valore di PR
srR = srR0i + om*srR1i; %calcolo srR
disp('stati corrispondenti')
v = Z*RT/P %calcolo V [m3/mol]
hr = hrRTC*RTc
sr = srR*R
gr = hr - T*sr
pause
%4 viriale
B0 = 0.083-0.422/TR^1.6; %calcolo B0
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
118
B1 = 0.139-0.172/TR^4.2; %calcolo B1
BPcRTc = B0+om*B1; %calcolo B
Z = 1+BPcRTc*PR/TR; %calcolo Z
dBdTRPc = 0.675/TR^2.6+om*0.722/TR^5.2;
hrRT =BPcRTc*PR/TR - dBdTRPc*PR;
srR =- dBdTRPc*PR;
disp('viriale')
v = Z*RT/P %calcolo V [m3/mol]
hr = hrRT*RT
sr = srR*R
gr = hr - T*sr
function [hrRT,srR] = fr(Z,A,B,STrk,tipo)
%fr calcola le funzioni residue con diverse EoS
% dati A, B, STrk=S*sqrt(TR/k)
% (calcolate coi dati di EoS.m),
% tipo (EoS da usare)
% tipo: 1 vdW
% 2 RK
% 3 RKS
% 4 PR
% restituisce hr/(RT) e sr/R
%
% uso: [hrRT,srR] = fr(Z,A,B,STrk,tipo)
% R. Rota (2002)
if tipo == 1 %vdW
hrRT =Z-1-A/Z;
srR =log(Z-B);
end
if tipo ==2 %RK
hrRT =Z-1-3*A/(2*B)*log((Z+B)/Z);
srR =log(Z-B)-A/(2*B)*log((Z+B)/Z);
end
if tipo ==3 %RKS
hrRT =Z-1-A/B*(1+STrk)*log((Z+B)/Z);
srR =log(Z-B)-A*STrk/B*log((Z+B)/Z);
end
if tipo == 4 %PR
hrRT =Z-1-A/(2*sqrt(2)*B)*(1+STrk)*...
log((Z+B*(1+sqrt(2)))/(Z+B*(1-sqrt(2))));
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
119
srR =log(Z-B)-A*STrk/(2*sqrt(2)*B)*...
log((Z+B*(1+sqrt(2)))/(Z+B*(1-sqrt(2))));
end
4.4 Diagrammi di stato
Come discusso nei paragrafi precedenti, dai valori di P - v - T calcolati mediante
unequazione di stato possibile calcolare le funzioni residue e quindi i valori delle
funzioni termodinamiche. Poich, come visto nel capitolo precedente, i valori sperimentali
P - v - T possono essere direttamente riportati su grafici o in tabelle, da tali dati possibile
risalire, tramite le relazioni discusse in precedenza, ai valori delle funzioni termodinamiche
che possono essere a loro volta riportate, per un dato fluido, in tabelle o grafici.
Questa procedura porta alla costruzione delle tabelle termodinamiche e dei diagrammi di
stato. Poich lo stato intensivo di un sistema omogeneo monocomponente completamente
caratterizzato dal valore di due variabili intensive, i vari tipi di diagrammi riportano oltre
alle due grandezze base sugli assi (per esempio P - v, T - s, h - s, ecc.) anche una serie di
curve parametrizzate su valori costanti di altre variabili, quali isobare (P = cost), isoterme
(T = cost), isotitolo (% di vapore costante nelle regioni bifasiche), isoentalpiche (h = cost),
isoentropiche (s = cost), ecc.
Il principale limite di questo approccio, analogamente al caso dei dati volumetrici, che i
diagrammi di stato sono disponibili solo per un numero limitato di fluidi estesamente
utilizzati, mentre lingegnere chimico si scontra quotidianamente con la necessit di
conoscere il valore delle funzioni termodinamiche dei fluidi pi disparati. Da questo
consegue lestrema importanza dei metodi di calcolo esposti nei paragrafi precedenti. Ci
non toglie che, quando sono disponibili per il fluido di interesse, i diagrammi di stato o le
tabelle termodinamiche forniscono un metodo semplice ed affidabile per il calcolo delle
funzioni termodinamiche.
4.4.1 Applicazione
Si scarica una bombola di azoto ben coibentata da 0,14 [m
3
] con una portata costante di 10
[mol min
-1
]. Inizialmente il gas si trova a 100 [atm] e 170 [K]. Calcolare il volume
specifico, la pressione e la temperatura del gas dopo 50 [min] col diagramma di stato
riportato in Figura 20 assumendo che lo scarico avvenga in modo reversibile.
Il numero di moli presente inizialmente si calcola facilmente una volta noto il volume
specifico (o la densit) nelle condizioni iniziali (100 [atm] e 170 [K]). Dal diagramma di
stato (punto 1) si ricava il valore ] m [kg 290 ] cm [g 29 , 0
3 3
= , da cui:
[kmol] 45 , 1
] kmol [kg 28
] m [kg 290 ] [m 14 , 0
1 -
-3 3
=
= =
M
V
n
i
(357)
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
120
Il bilancio di materia fornisce una relazione tra il numero di moli presenti allinizio e quelle
presenti dopo un certo tempo:
t n n n dt n dn n
dt
dn
i f
t n
n
f
i
& & & = = =
0
(358)
Dopo 50 [min] sono presenti nella bombola
[kmol] 95 , 0 50 010 , 0 45 , 1 = =
f
n (359)
Il volume specifico nelle condizioni finali quindi pari a:
] kmol [m 147 , 0
[kmol] 95 , 0
] [m 14 , 0
1 - 3
3
= = =
f
f
n
V
v (360)
e quindi la densit massica :
] m [kg 5 , 190
] kmol [m 147 , 0
] kmol [kg 28
3
1 - 3
-1
= = =
f
f
v
M
(361)
Poich lo scarico avviene in modo adiabatico e reversibile, la trasformazione del gas
isoentropica. Sul diagramma di stato, partendo dal punto che caratterizza le condizioni
iniziali (punto 1: P = 100 [atm] ~ 10 [MPa] e T = 170 [K] ~ 153 [F]) si scende lungo una
isoentropica fino a incrociare la curva con densit pari a 190 [kg m
-3
] ~ 12 [lb ft
-3
] (punto 2
sul diagramma di stato). In corrispondenza del punto 2 si legge una temperatura di circa
210 [F] ~140 [K] e una pressione di circa 40 [bar].
4.5 Fasi condensate
Come discusso in precedenza, luso di equazioni di stato in grado di rappresentare il
comportamento volumetrico anche della fase liquida consente il calcolo delle funzioni
residue, e quindi del valore dellentalpia e dellentropia, anche per la fase liquida. Si
anche detto in precedenza che le equazioni di stato, sviluppate come correzioni sempre pi
accurate dellEoS del gas perfetto, sono generalmente in grado di rappresentare meglio il
comportamento della fase gas rispetto a quello della fase liquida. Di conseguenza, anche il
valore delle funzioni residue calcolate con le relazioni discusse nella sezione precedente
sono pi affidabili per la fase gas che non per la fase liquida.
Un approccio alternativo (solitamente detto metodo indiretto) per il calcolo delle funzioni
termodinamiche delle fasi condensate che non richiede lutilizzo delle equazioni di stato per
la fase condensata quello che sfrutta unulteriore informazione sperimentale: il calore
latente di transizione di fase (cio, di evaporazione o di fusione).
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
121
Figura 20: diagramma di stato dellazoto.
e
n
t
a
l
p
i
a
[
k
J
k
g
-
1
]
pressione [MPa]
1
2
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
122
Poich le transizione di fase sono isotermobariche, il calore latente associato alla
transizione di fase coincide, per la relazione (62), con la variazione di entalpia tra le due
fasi, mentre la variazione di entropia coincide col calore latente diviso per la temperatura a
cui avviene la transizione di fase:
( ) ( ) ( ) ( ) ( )
( ) ( ) ( ) ( ) ( )
( ) ( ) ( )
( ) ( )
( ) ( ) ( )
( ) ( )
fus
fus fus
fus
fus fus
fus fus fus
Ss
fus
Ls
ev
ev ev
ev
ev ev
ev ev ev
Ls
ev
Vs
fus solid fus fus fus fus fus
Ss
fus
Ls
ev cond ev ev ev ev ev
Ls
ev
Vs
T
T h
T
T q
T s T s T s
T
T h
T
T q
T s T s T s
T q T q T h T h T h
T q T q T h T h T h
= = =
= = =
= = =
= = =
(362)
dove gli apici indicano il vapore saturo (V
s
), il liquido saturo (L
s
, inteso come liquido in
equilibrio col vapore o col solido a seconda dei casi) e il solido saturo (S
s
, cio in equilibrio
col liquido). I pedici invece indicano le condizioni di evaporazione (ev), condensazione
(cond), fusione (fus) e solidificazione (solid), mentre con q si indicato il calore latente
specifico associato alla transizione di fase. Tutte le grandezze dipendono solo dalla
temperatura in quanto, essendo in condizioni di equilibrio tra due fasi, data la temperatura
risulta fissata in modo univoco la pressione (pari alla tensione di vapore, nel caso di
evaporazione, e alla pressione di fusione, in caso di fusione, alla temperatura data).
Utilizzando questi dati, possibile calcolare lentalpia specifica di un composto puro in
fase liquida come segue
70
:
( ) ( ) ( ) ( )
( ) ( ) ( )
+ + + =
= + = +
T
T
L
P ev ev ev
Vs R
T
T
P r
T
T
L
P ev ev ev
Vs
T
T
L
P ev
Ls L
ev
ev
r
ev ev
dT c T h P T h dT c T h
dT c T h T h dT c T h P T h
,
,
, * *
(363)
dove con T
ev
si indicata la temperatura di equilibrio liquido - vapore alla pressione
considerata, P.
Si nota che in questa relazione la grandezza residua viene calcolata per la fase vapore
invece che per quella liquida, con una maggiore affidabilit. Il prezzo da pagare la
conoscenza di alcune informazioni sperimentali in pi, lentalpia di evaporazione e il calore
70
In queste relazioni si trascurata la dipendenza di h e s dalla pressione, approssimazione
solitamente lecita per le fasi condensate lontano dal punto critico e per salti di pressione non troppo
elevati.
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
123
specifico del liquido. La relazione tra tali grandezze sperimentali e le funzioni residue viene
dedotta confrontando la relazione precedente con la (276):
( ) ( ) ( )
( ) ( ) P T h dT c T h
dT c T h P T h dT c T h
L R
T
T
P r
T
T
L
P ev ev ev
Vs R
T
T
P r
r
ev
ev
r
,
,
, * *
, * *
+ + =
= + + +
(364)
Se si considera T = T
ev
, possibile ricavare una semplice relazione tra i valori delle
funzioni residue del liquido e del vapore saturo e lentalpia di evaporazione:
( ) ( ) ( )
ev ev ev
Ls R
ev
Vs R
T h P T h P T h = , ,
, ,
(365)
Analogamente, il calcolo dellentropia di un composto puro in fase liquida pu essere fatto
come:
( ) ( ) ( ) ( )
( ) ( )
( )
+ +
|
|
.
|
\
|
+ =
= + = +
T
T
L
P
ev
ev ev
ev
Vs R
r
T
T
P
r r
T
T
L
P
ev ev ev
Vs
T
T
L
P
ev
Ls L
ev
ev
r
ev ev
dT
T
c
T
T h
P T s
P
P
R dT
T
c
P T s
dT
T
c
T s T s dT
T
c
T s P T s
, ln ,
,
,
*
*
(366)
Analogamente al caso dellentalpia,:
( ) ( )
( )
( ) ( ) P T s
P
P
R dT
T
c
P T s
dT
T
c
T
T h
P T s
P
P
R dT
T
c
P T s
L R
r
T
T
P
r r
T
T
L
P
ev
ev ev
ev
Vs R
r
T
T
P
r r
r
ev
ev
r
, ln ,
, ln ,
,
*
*
,
*
*
+
|
|
.
|
\
|
+ =
= +
+
|
|
.
|
\
|
+
(367)
Se si considera T = T
ev
, ancora possibile ricavare una semplice relazione tra i valori delle
funzioni residue del liquido e del vapore saturo e lentalpia di evaporazione:
( ) ( )
( )
ev
ev ev
ev
Ls R
ev
Vs R
T
T h
P T s P T s
= , ,
, ,
(368)
Analogamente, lentalpia e lentropia specifiche di un solido puro possono essere calcolate
come:
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
124
( ) ( ) ( ) ( )
+ = +
T
T
S
P fus fus fus
Ls
T
T
S
P fus
Ss S
fus fus
dT c T h T h dT c T h P T h ,
(369)
( ) ( ) ( ) ( )
+ = +
T
T
S
P
fus fus fus
Ls
T
T
S
P
fus
Ss S
fus fus
dT
T
c
T s T s dT
T
c
T s P T s ,
(370)
dove con T
fus
si indicata la temperatura di equilibrio liquido - solido alla pressione
considerata, P. Lentalpia e lentropia specifica del liquido saturo (in questo caso, in
equilibrio col solido) si calcolano coi metodi visti in precedenza.
4.5.1 Applicazione
Si calcoli, con lEoS PR, la differenza di entalpia ed entropia del n-ottano tra lo stato 1: fase
vapore a 427.9 [K] e 2.15 [bar], e lo stato 2: fase liquida a 427.9 [K] e 5 [bar]
71
.
Si tratta di calcolare le grandezze termodinamiche nei due stati e farne poi la differenza.
Utilizzando la relazione (278) si ottiene:
( ) ( )
1 1
,
2 2
, *
1 1 2 2 2 1
, , ) , ( ) , (
2
1
P T h P T h dT c P T h P T h h
V R L R
T
T
P
V L
+ = =
(371)
NellApplicazione 4.3.4 si sono gi calcolati, con lEoS PR, i valori di
( ) ] mol [J 33363 ,
1
2 2
,
= P T h
L R
e ( ) ] mol [J 890 ,
1
1 1
,
= P T h
V R
. Come riportato nella
Tabella 3, il calore specifico a pressione costante del n-ottano come gas perfetto pu essere
calcolato dalla relazione (T in [K]):
2 6 3 *
10 208 , 22 10 567 , 70 163 , 8 T T R c
P
+ = . La
variazione di entalpia specifica si calcola quindi come:
( )
( ) ( ) ] mol [J 32473 890 33363
10 208 , 22 10 567 , 70 163 , 8 314 , 8
1
9 , 427
9 , 427
2 6 3
2 1
= +
+ + =
dT T T h
(372)
Analogamente, il calcolo della variazione di entropia specifica , dalla relazione (279):
71
Si noti che il quesito pu essere riformulato nel seguente modo equivalente: si calcoli lentalpia e
lentropia del n-ottano in fase vapore a 427.9 [K] e 2.15 [bar] assumendo come stato di riferimento la
fase liquida a 427.9 [K] e 5 [bar].
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
125
( ) ( )
( ) ( ) ] K mol [J 90 , 75 40 , 1 28 , 70
15 , 2
5
ln 314 , 8
10 208 , 22 10 567 , 70 163 , 8
314 , 8
, , ln
) , ( ) , (
1 1
9 , 427
9 , 427
2 6 3
1 1
,
2 2
,
1
2
*
1 1 2 2 2 1
2
1
= +
|
|
.
|
\
|
+
|
|
.
|
\
|
+
=
= +
|
|
.
|
\
|
=
= =
dT
T
T T
P T s P T s
P
P
R dT
T
c
P T s P T s s
V R L R
T
T
P
V L
(373)
Lo stesso calcolo pu essere ripetuto con il metodo indiretto (i.e., equazioni (363) e (366)),
conoscendo la temperatura di ebollizione normale
72
(T
ev
= 398,8 [K]), lentalpia di
evaporazione normale (h
ev
= 34,799 [kJ mol
-1
]) e il calore specifico del liquido ( 330 =
L
P
c
[J mol
-1
K
-1
]):
( ) ( ) ( )
( ) ( )
( ) ( ) ( )
( ) ( )
( ) ] mol [J 33163 890 330
34799 452 10 208 , 22 10 567 , 70 163 , 8 314 , 8
,
,
) , ( ) , (
1
9 , 427
8 , 398
8 , 398
9 , 427
2 6 3
1 1
, , *
1 1
, * *
, * *
1 1 2 2 2 1
2
1
1
2
= +
+ + + =
= + + =
=
(
+ +
+
(
(
+ + + =
= =
dT
dT T T
P T h dT c T h T h dT c
P T h dT c T h
dT c T h T h dT c T h
P T h P T h h
V R
T
T
L
P ev ev ev
Vs R
T
T
P
V R
T
T
P r
T
T
L
P ev ev ev
Vs R
T
T
P r
V L
ev
ev
r
ev
ev
r
(374)
Il valore dellentalpia residua del vapore saturo a pressione ambiente (pari a - 452 [J mol
-1
])
stato calcolato col programma vhsgTP.m illustrato nella Applicazione 4.3.4.
Analogamente si pu calcolare lentropia specifica:
72
Col termine normale si intende a pressione atmosferica.
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
126
( ) ( )
( )
( ) ( )
( )
( )
( )
( ) ( )
] K mol [J 64 , 76
40 , 1
330
8 , 398
34799
75 , 0
15 , 2
1
ln 314 , 8
10 208 , 22 10 567 , 70 163 , 8
314 , 8
, ln
, ln ,
ln ,
) , ( ) , (
1 1
9 , 427
8 , 398
8 , 398
9 , 427
2 6 3
1 1
, ,
1
*
1 1
, 1
*
*
,
*
*
1 1 2 2 2 1
2
1
1
2
=
= + +
|
|
.
|
\
|
+
|
|
.
|
\
|
+
=
= +
+
|
|
.
|
\
|
=
=
(
(
+
|
|
.
|
\
|
+
+
(
(
+
|
|
.
|
\
|
+ =
= =
dT
T
dT
T
T T
P T s dT
T
c
T
T h
T s
P
P
R dT
T
c
P T s
P
P
R dT
T
c
P T s
dT
T
c
T
T h
T s
P
P
R dT
T
c
P T s
P T s P T s s
V R
T
T
L
P
ev
ev ev
ev
Vs R ev
T
T
P
V R
r
T
T
P
r r
T
T
L
P
ev
ev ev
ev
Vs R
r
ev
T
T
P
r r
V L
ev
ev
r
ev
ev
r
(375)
Il valore dellentropia residua del vapore saturo a pressione ambiente (pari a - 0,75 [J mol
-1
K
-1
]) stato calcolato col programma vhsgTP.m illustrato nella Applicazione 4.3.4.
4.6 Propriet termodinamiche in condizione di equilibrio di
fase
Quando in un sistema monocomponente sono presenti pi di una fase, le condizioni di
equilibrio termico e meccanico discusse nella sezione 1.4 richiedono che la temperatura e la
pressione siano uguali in tutte le fasi. Inoltre, affinch il sistema sia in equilibrio anche
rispetto al trasferimento di materia tra le fasi necessario che il potenziale chimico del
composto in tutte le fasi assuma lo stesso valore. Considerando per esempio un sistema
monocomponente contenente una fase liquida e una fase vapore in equilibrio deve essere
vero che:
( ) ( ) = P T P T
V L
, , (376)
dove si indicato con P la tensione di vapore del composto alla temperatura T, cio
lunico valore di pressione per cui, allassegnata temperatura, possono coesistere le due fasi
in equilibrio.
Il potenziale chimico ha un legame immediato con lenergia libera di Gibbs. Infatti,
considerando un sistema monocomponente e monofase e ricordando che lenergia libera di
Gibbs una funzione di stato, la relazione (238) diventa:
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
127
dn
n
G
dV
P
G
dT
T
G
dn VdP SdT dG
P T n T n P , , ,
|
.
|
\
|
+ |
.
|
\
|
+ |
.
|
\
|
= + + =
(377)
da cui si deducono le relazioni:
= |
.
|
\
|
= |
.
|
\
|
= |
.
|
\
|
P T
n T
n P
n
G
V
P
G
S
T
G
,
,
,
(378)
Dallultima di queste relazioni si ottiene poi la relazione cercata tra il potenziale chimico e
lenergia libera di Gibbs molare:
g
n
n
g
n
ng
n
G
P T P T P T
= |
.
|
\
|
= |
.
|
\
|
= |
.
|
\
|
=
, , ,
(379)
dove si sfruttato il fatto che se sono fissate due variabili intensive (T e P, in questo caso)
anche tutte le altre (in particolare g) sono costanti. Limportante conclusione quindi che
per un composto puro il potenziale chimico coincide con lenergia libera di Gibbs molare e
la relazione (376) diventa:
( ) ( ) = P T g P T g
V L
, , (380)
Un sistema costituito da una fase liquida e da una fase vapore in equilibrio alla temperatura
T
1
e alla pressione P(T
1
) rappresentato dal punto 1 nella Figura 21.
Se si aumenta la temperatura del sistema di una quantit infinitesima dT mantenendo la
presenza delle due fasi in equilibrio, la tensione di vapore deve aumentare di una quantit
infinitesima dP: il sistema si porta quindi al punto 2 di Figura 21. Sia nel punto 1 sia nel
punto 2 deve valere la relazione (380):
( ) ( ) ( ) ( )
( ) ( ) ( ) ( ) dP T P dT T g dP T P dT T g
T P T g T P T g
V L
V L
+ + = + +
=
1 1 1 1
1 1 1 1
, ,
, ,
(381)
Poich:
( ) ( ) ( ) ( )
( ) ( ) ( ) ( )
V V V
L L L
dg T P T g dP T P dT T g
dg T P T g dP T P dT T g
+ = + +
+ = + +
1 1 1 1
1 1 1 1
, ,
, ,
(382)
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
128
ne consegue che nella trasformazione dallo stato 1 allo stato 2 la variazione infinitesima
della energia libera di Gibbs specifica della fase vapore deve essere uguale a quella della
fase liquida:
V L
dg dg = (383)
Figura 21: diagramma di stato di un composto puro.
Utilizzando la relazione (240) si ottiene:
dP v dT s dg dP v dT s dg
V V V L L L
+ = = + = (384)
da cui:
curva di fusione per composti che si
espandono solidificando
P
[
b
a
r
]
T [K]
vapore
liquido
curva di
evaporazione
curva di
sublimazione
punto critico
T
1
+dT
solido
punto
triplo
T
1
P(T
1
)
dT
1
2
P(T
1
)+dP
dP
curva di fusione per composti
che si espandono fondendo
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
129
( )
ev
ev
L V
L V
L V
L V
v T
h
v v
T h h
v v
s s
dT
dP
= (385)
Questa relazione, che fornisce la dipendenza della tensione di vapore dalla temperatura
attraverso il calore latente di evaporazione (
ev
h ) e la variazione di volume molare nella
transizione di fase (
L V
ev
v v v = ) nota come equazione di Clapeyron
73
.
Relazioni analoghe possono essere dedotte anche per le altre transizioni di fase, tra liquido
e solido e tra vapore e solido, pur di inserire nella relazione (385) il calore latente e la
variazione di volume molare relativi alla transizione di fase considerata.
Poich i calori latenti di evaporazione, fusione e sublimazione sono sempre positivi, la
pendenza (dP/dT) delle curve di coesistenza di due fasi sul diagramma di Figura 21
dipendono dal segno di v . La variazione di volume da liquido a vapore e da solido a
vapore sempre positiva e quindi la pendenza delle curve di evaporazione e di
sublimazione sempre positiva. In altri termini, la tensione di vapore del liquido e del
solido aumentano sempre con la temperatura. Per molti composti 0 >
fus
v , cio il volume
molare del liquido maggiore di quello del solido in equilibrio. Per questi composti dP/dT
positivo anche per il ramo di equilibrio liquido - solido e la temperatura di fusione
aumenta con la pressione. Viceversa, per composti come lacqua il cui volume molare
liquido minore di quello solido la pendenza della curva di fusione negativa e la
temperatura di fusione diminuisce se la pressione aumenta
74
.
Considerando la curva di evaporazione a pressioni non troppo elevate e lontano dal punto
critico, possibile approssimare la relazione (385) considerando il volume molare del gas
molto maggiore di quello del liquido e simile a quello di un gas perfetto:
P
RT
v v v v
V L V
ev
= (386)
Inserendo questa approssimazione nella relazione (385) si ottiene la equazione di Clausius -
Clapeyron:
( )
2 2
ln
RT
h
dT
P d
RT
h P
dT
dP
ev ev
=
=
(387)
73
Benoit Paul mile Clapeyron, 1799 1864, fisico e ingegnere francese.
74
Questo fatto ha molte interessanti implicazioni: per esempio, il giaccio galleggia sullacqua e forma
uno strato superficiale sui corpi idrici. Quando una persona pattina sul ghiaccio esercita una certa
pressione sul ghiaccio stesso provocandone quindi una riduzione della temperatura di fusione. Questo
provoca la fusione del ghiaccio con la formazione di un film di acqua tra la lama del pattino e la
superficie solida che agisce come lubrificante, rendendo scivoloso il ghiaccio.
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
130
Questa relazione pu essere utilizzata per stimare il valore del calore latente di
evaporazione da due valori di tensione di vapore, oppure per dedurre landamento
qualitativo della tensione di vapore con la temperatura.
Integrando tra due valori di temperatura (trascurando la variazione del calore latente con la
temperatura) si ottiene:
( )
( )
( ) ( ) ( )
( )
2 1
1 2
2 1 1
2
1 1
ln 1 1
ln
T T
T P T P R
h
T T R
h
T P
T P
ev
ev
=
|
|
.
|
\
|
=
|
|
.
|
\
|
(388)
Integrando in modo indefinito si ottiene invece:
( ) ( )
T
B
A P
RT
h
P
ev
+ = +
= ln cost ln (389)
che mostra come i valori del logaritmo della tensione di vapore in funzione dellinverso
della temperatura si devono allineare su di una retta. Questo andamento ben verificato
sperimentalmente per molti composti a bassa pressione e fornisce la base per la seguente
relazione semiempirica, detta equazione di Antoine
75
, estesamente utilizzata per prevedere
la tensione di vapore in funzione della temperatura:
( )
T C
B
A P
+
+ = ln (390)
Le costanti A, B e C, caratteristiche di ciascun composto, sono facilmente reperibili nei
manuali tecnici
76
.
Il calore latente di evaporazione anchesso disponibile per molte sostanze nei manuali
tecnici. Alla temperatura normale di ebollizione il calore latente di evaporazione pu essere
stimato con un errore dellordine del 2 % con la relazione di Riedel
77
:
( )
( )
|
|
.
|
\
|
=
R bn
C
R bn C bn ev
T
P
T RT T h
,
,
930 , 0
013 , 1 ln
093 , 1
(391)
75
Ch. Antoine, Comtes Rendus de lAcadmie de Science, 107, 681 (1888).
76
Si veda per esempio: K.C. Reid, J.M. Prausnitz, B.E. Poling The properties of gases and liquids,
McGraw-Hill, N.Y. (1988). Lutilizzo di queste costanti richiede una certa cautela, in quanto esse
dipendono dalla particolare formulazione dellequazione di Antoine riportata in un dato manuale. In
particolare, il logaritmo pu essere sia naturale sia decimale e le unit di misura della tensione di
vapore e della temperatura possono essere le pi diverse.
77
L. Riedel, Chem. Ing. Tech., 26, 679 (1954).
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
131
dove T
C
[K] e P
C
[bar] sono la temperatura e la pressione critica, e T
bn,R
= T
bn
/T
C
la
temperatura di ebollizione normale ridotta. La dipendenza del calore latente di
evaporazione dalla temperatura pu poi essere stimata con la relazione di Watson
78
:
( )
( )
38 , 0
2
1
2
1
1
1
|
|
.
|
\
|
R
R
ev
ev
T
T
T h
T h
(392)
dove T
R
la temperatura ridotta.
4.7 Fugacit
Come discusso in precedenza, il potenziale chimico la variabile termodinamica che
fornisce il criterio per lequilibrio al trasferimento di materia. Il potenziale chimico di un
composto puro coincide con lenergia libera di Gibbs molare che, per un gas perfetto, pu
essere calcolata integrando la relazione (240). A temperatura costante questa relazione
fornisce:
( ) P RTd dP
P
RT
dP v dg d
T T
ln
* * *
= = = = (393)
che integrata tra due valori di pressione diventa:
( ) ( )
|
|
.
|
\
|
=
2
1
2
*
1
*
ln , ,
P
P
RT P T P T (394)
Questa relazione consente di calcolare la differenza di potenziale chimico tra due sistemi
costituiti da un gas perfetto in funzione di una variabile misurabile, la pressione.
Purtroppo si tratta di un risultato poco utile nella pratica, in quanto si spesso interessati al
trasferimento di materia tra due fasi di cui una necessariamente condensata e quindi
sicuramente non assimilabile a un gas perfetto. Daltro canto rimane la necessit di poter
correlare il potenziale chimico a delle variabili misurabili per definire le condizioni di
equilibrio al trasferimento di materia, in modo analogo a quanto fatto nella sezione
precedente per dedurre lequazione di Clapeyron. Questo storicamente stato fatto da
Lewis
79
introducendo una nuova variabile termodinamica chiamata fugacit e solitamente
indicata con f. Questa variabile viene definita (per fluidi qualsiasi, assimilabili o meno a un
gas perfetto) in termini differenziali per analogia alla relazione (393) come:
( ) f RTd d
T
ln = (395)
78
K.M. Watson, Ind. Eng. Chem., 35, 398 (1943).
79
Gilbert Newton Lewis, 1857 - 1946, chimico - fisico statunitense.
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
132
Questa definizione consente solo il calcolo della differenza di fugacit. Per poterne
calcolare il valore assoluto quindi necessario definirne il valore in un punto. Poich
confrontando le relazioni (393) e (395) risulta evidente che la fugacit di un composto puro
come gas perfetto coincide con la pressione e ricordando che tutti i composti per pressioni
sufficientemente basse si comportano come gas perfetti, la definizione (395) viene
completata dalla seguente:
1
lim
0
= |
.
|
\
|
P
f
P
(396)
Per un composto puro, il rapporto tra la fugacit e la pressione viene solitamente chiamato
coefficiente di fugacit, , che pu anche essere interpretato come il rapporto tra la fugacit
di un fluido reale e quella di un gas perfetto nelle stesse condizioni:
( )
( )
( )
( )
P
P T f
P T f
P T f
P T
,
,
,
,
*
= =
(397)
La relazione (396) diviene quindi:
( ) 1
lim
0
=
P
(398)
Risulta evidente che il coefficiente di fugacit di un gas perfetto uguale a uno.
4.7.1 Condizioni di equilibrio
La condizione di equilibrio al trasferimento di materia, per esempio tra una fase liquida e
una fase vapore, pu essere espressa in funzione delle fugacit integrando la relazione (395)
tra le due fasi:
( ) ( ) ( )
( )
( )
|
|
.
|
\
|
= =
P T f
P T f
RT P T P T f RTd d
L
V
L V
V P T
L P T
V P T
L P T
T
,
,
ln , , ln
, ,
, ,
, ,
, ,
(399)
In condizioni di equilibrio P = P(T) e i due potenziali chimici devono essere uguali. Ne
consegue che in tali condizioni anche la fugacit del composto nelle due fasi deve essere
uguale:
( ) ( ) ( ) ( ) T P T f T P T f
L V
= , , (400)
Questa rappresenta una condizione di equilibrio del tutto analoga alluguaglianza dei
potenziali chimici e, sfruttando la definizione di coefficiente di fugacit, pu anche essere
posta nella forma equivalente:
( ) ( ) ( ) ( ) ( ) ( ) ( ) ( ) ( ) ( ) T P T T P T T P T T P T P T T P
L V L V
= = , , , , (401)
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
133
Perch questa relazione sia operativa per necessario definire una procedura per il calcolo
del coefficiente di fugacit (o, equivalentemente, della fugacit) sulla base di grandezze
misurabili.
4.7.2 Calcolo della fugacit con equazioni di stato
Sottraendo la relazione (393) dalla (395) si ottiene:
( ) ( ) ln ln
* * *
RTd
P
f
RTd dg g g d dg dg d d
R
T T T T T
= |
.
|
\
|
= = = = (402)
Integrando questa relazione a temperatura e pressione costante tra lo stato di gas perfetto
(dove per definizione g
R
= 0 e = 1) e quello di fluido reale (sia liquido sia vapore) si
ottiene:
( ) ( )
RT
g
d
RT
dg
R
P T
P T
P T
P T
R
= =
ln ln
,
,* ,
,
,* ,
(403)
Questa relazione consente il calcolo del coefficiente di fugacit utilizzando le relazioni gi
sviluppate per il calcolo dellenergia libera di Gibbs residua.
In particolare, utilizzando lEoS del viriale il coefficiente di fugacit risulta dalla relazione:
( )
RT
PB
RT
g
R
= = ln
(404)
mentre utilizzando le EoS cubiche il coefficiente di fugacit risulta dalle relazioni riassunte
in Tabella 10, riprodotte per chiarezza nella Tabella 12.
EoS ln()
vdW ( ) B Z
Z
A
Z ln 1
RK ( ) B Z
Z
B Z
B
A
Z |
.
|
\
| +
ln ln 1
RKS ( ) B Z
Z
B Z
B
A
Z |
.
|
\
| +
ln ln 1
PR
( )
( )
( ) B Z
B Z
B Z
B
A
Z
|
|
.
|
\
|
+
+ +
ln
2 1
2 1
ln
2 2
1
Tabella 12: espressioni per il calcolo del coefficiente di fugacit di composti puri con
alcune EoS cubiche.
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
134
Poich le equazioni di stato cubiche sono in grado di rappresentare il comportamento della
fase vapore e di quella liquida, possibile utilizzarle per calcolare il coefficiente di fugacit
di entrambe le fasi. La procedura di calcolo in questo caso la seguente:
assegnare temperatura e pressione e calcolare i parametri A e B;
risolvere la cubica in Z e selezionare la radice minore (se la pressione maggiore della
tensione di vapore: fase liquida) o quella maggiore (se la pressione minore della
tensione di vapore: fase vapore);
calcolare il coefficiente di fugacit con la relazione opportuna.
Anche la relazione per il calcolo della energia libera di Gibbs residua (273) pu essere
poste in una forma generalizzata inserendo le definizioni (302):
( ) ( ) ( ) cost 1 1 ln
0 0
= = = =
T
P
dP
Z
P
dP
Z
RT
g
R
P
R
R
P
R
(405)
Si nota che il coefficiente di fugacit dipende solo dalla pressione ridotta e dal coefficiente
di compressibilit, Z. Utilizzando la legge degli stati corrispondenti a tre parametri (171) si
ottiene:
( ) ( ) ( )
( ) ( )
cost
ln ln
1 1 ln
1 0
1 0 1 0
=
+ =
+ = + =
T
P
dP
Z
P
dP
Z
P
dP
Z Z
P
o
R
R
P
o
R
R
P
o
R
R
(406)
I due integrali possono essere valutati in funzione delle sole variabili T
R
e P
R
dai dati
tabulati di Z
0
(T
R
,P
R
) e Z
1
(T
R
,P
R
). I due gruppi ( ) ( )
1 0
ln e ln dipendono quindi solo dalle
variabili ridotte e possono essere riportati in grafici o tabelle analogamente a quanto fatto
per le due funzioni Z
0
(T
R
,P
R
) e Z
1
(T
R
,P
R
). I valori proposti per queste funzioni da Lee e
Kesler sono riportati in Appendice C.
Analogamente al caso del coefficiente di compressibilit, la funzione ( )
1
ln rappresenta
una correzione ai valori della funzione ( )
0
ln e pu quindi essere spesso trascurata per una
valutazione di massima.
4.7.3 Fasi condensate
Luso di equazioni di stato in grado di rappresentare il comportamento volumetrico anche
della fase liquida consente il calcolo della fugacit anche per la fase liquida. Daltro canto,
le equazioni di stato sono generalmente in grado di rappresentare meglio il comportamento
della fase gas rispetto a quello della fase liquida. Di conseguenza, anche il valore della
fugacit calcolato con le relazioni discusse nella sezione precedente sono pi affidabili per
la fase gas che non per la fase liquida.
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
135
Un approccio alternativo per il calcolo della fugacit delle fasi condensate che non richiede
lutilizzo delle equazioni di stato per la fase condensata (solitamente detto metodo indiretto)
quello che sfrutta unulteriore informazione sperimentale: la tensione di vapore.
Lapproccio concettualmente analogo a quello discusso nella sezione 4.5 per il calcolo
delle funzioni residue.
In condizioni di equilibrio liquido - vapore la fugacit del composto puro nelle due fasi
deve essere uguale. Inoltre, a una data temperatura le due fasi coesistono in equilibrio a un
solo valore pressione: la tensione di vapore alla temperatura assegnata. Quindi le condizioni
di equilibrio sono rappresentate dalla relazione (400):
( ) ( ) ( ) ( ) ( ) ( ) ( ) T P T T P T P T f T P T f
V V L
= = , , , (407)
Quindi la fugacit del composto in fase liquida alla pressione P(T) pu essere calcolata
moltiplicando la tensione di vapore per il coefficiente di fugacit della fase vapore.
Volendo calcolare il valore della fugacit a una diversa pressione (ma alla stessa
temperatura) si pu sfruttare la definizione di fugacit (395) insieme alla relazione (240).
Ricordando che per un composto puro il potenziale chimico coincide con lenergia libera di
Gibbs molare, a temperatura costante si ottiene che:
( ) vdP dg f RTd d
T T
= = = ln (408)
Integrando tra la tensione di vapore e una pressione generica per un composto in fase
liquida:
( )
=
L P T
L T P T
L
L P T
L T P T
L
dP v f RTd
, ,
), ( ,
, ,
), ( ,
ln
(409)
si ottiene la relazione cercata:
( )
( ) ( )
=
|
|
.
|
\
|
L P T
L T P T
L
L
L
dP v
T P T f
P T f
RT
, ,
), ( ,
,
,
ln
(410)
che fornisce la dipendenza della fugacit dalla pressione:
( ) ( ) ( )
|
|
.
|
\
|
=
L P T
L T P T
L
L L
dP
RT
v
T P T f P T f
, ,
), ( ,
exp , , (411)
Lesponenziale presente in questa relazione prende il nome di correzione di Poynting
80
. In
condizioni lontane da quelle critiche e per variazioni di pressione non troppo elevate
80
John Henry Poynting, 1852 - 1914, fisico inglese.
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
136
possibile considerare costante il volume specifico delle fasi condensate e la relazione
precedente diventa (inserendo anche la relazione (407)):
( ) ( ) ( )
( )
( ) ( ) ( )
( )
|
|
.
|
\
|
=
=
|
|
.
|
\
|
RT
T P P v
T P T T P
RT
T P P v
T P T f P T f
L
V
L
L L
) (
exp ,
) (
exp , ,
(412)
Questa relazione consente di calcolare la fugacit del liquido a qualsiasi temperatura e
pressione senza utilizzare lequazione di stato per la fase condensata.
Poich il valore numerico del volume molare dei liquidi solitamente molto piccolo, la
correzione di Poynting normalmente trascurabile per variazioni di pressione modeste.
Questo conferma la regola generale che le propriet delle fasi condensate, e quindi anche la
fugacit, dipendono poco dalla pressione. Inoltre, se il fluido in fase gas a T e P(T) si
comporta come un gas perfetto il suo coefficiente di fugacit unitario e quindi la fugacit
del liquido risulta circa uguale alla tensione di vapore:
( ) ( ) T P P T f
L
, (413)
Lo stesso procedimento pu essere utilizzato per il calcolo della fugacit di un composto
solido sfruttando le condizioni di equilibrio solido - vapore, e quindi la tensione di vapore
del solido. In questo caso la relazione finale approssimata (trascurando la correzione di
Poynting e approssimando il comportamento del vapore a quello del gas perfetto) diventa:
( ) ( ) T P P T f
S S
, (414)
dove P
S
(T) rappresenta la tensione di vapore del solido.
Questo approccio risulta in realt utile per condizioni in cui la tensione di vapore del solido
non sia trascurabile, come invece avviene per la maggior parte dei solidi a temperature
lontane da quelle di fusione alla pressione considerata. In questo caso risulta pi utile un
approccio ancora diverso per calcolare la fugacit del composto in fase solida. Integrando la
relazione (395) tra la fase solida e quella liquida alla stessa temperatura e pressione:
( )
=
S P T
L P T
S P T
L P T
T
f RTd d
, ,
, ,
, ,
, ,
ln
(415)
si ottiene la seguente relazione:
( ) ( )
( )
( )
|
|
.
|
\
|
=
P T f
P T f
RT P T P T
L
S
L S
,
,
ln , , (416)
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
137
Poich il potenziale chimico per un composto puro coincide con lenergia libera di Gibbs
molare la relazione precedente diventa:
( )
( )
( ) ( ) ( )
( )
|
|
.
|
\
|
=
=
|
|
.
|
\
|
=
|
|
.
|
\
|
=
RT
P T g
RT
P T g
RT
P T g P T g
P T f
P T f
fus
sol
L S
L
S
,
exp
,
exp
, ,
exp
,
,
(417)
La variazione di energia libera di Gibbs molare legata alla transizione di stato da liquido a
solido (solidificazione, indicata col pedice
sol
, mentre la trasformazione inversa, fusione,
indicata col pedice
fus
) a una data temperatura e pressione pu essere calcolata come:
( ) ( ) ( ) P T s T P T h P T g
sol sol sol
, , , = (418)
Lentalpia e lentropia molare di solidificazione a (T, P) possono essere calcolate mediante
il ciclo immaginario schematizzato in Figura 22.
Figura 22: ciclo immaginario per il calcolo dellentalpia e dellentropia molare di
solidificazione a (T, P). T
f
la temperatura di fusione alla pressione P.
Poich sia lentalpia sia lentropia sono funzioni di stato la loro variazione dipende solo
dallo stato iniziale e finale e non dalla trasformazione seguita per passare da uno stato
allaltro. Ne consegue che, con riferimento alla Figura 22:
T
T
f
T
L
S
fase
d
c b
a
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
138
( )
d c c b b a d a sol
h h h h P T h
+ + = = , (419)
Le tre differenze di entalpia specifica possono essere calcolate da valori misurabili
81
:
( )
( )
f
S
P
T
T
S
P d c
f fus f sol c b
f
L
P
T
T
L
P b a
T T c dT c h
T h T h h
T T c dT c h
f
f
=
= =
=
) ( ) (
(420)
Analogamente, la variazione di entropia pu essere calcolata come:
|
|
.
|
\
|
=
=
|
|
.
|
\
|
=
f
S
P
T
T
S
P
d c
f
f fus
f
f sol
c b
f L
P
T
T
L
P
b a
T
T
c dT
T
c
s
T
T h
T
T h
s
T
T
c dT
T
c
s
f
f
ln
) ( ) (
ln
(421)
Ne consegue che:
( ) ( )
|
|
.
|
\
|
|
|
.
|
\
|
+
|
|
.
|
\
|
=
T
T
T T T c c
T
T
h P T g
f
f
S
P
L
P
fi
fus sol
ln 1 , (422)
e quindi la fugacit di un composto in fase solida risulta calcolabile come:
( ) ( )
|
|
.
|
\
|
|
|
.
|
\
|
|
|
.
|
\
|
+
|
|
.
|
\
|
=
T
T
T
T
R
c c
T T R
h
P T f P T f
f f
S
P
L
P
f
fus
L S
ln 1
1 1
exp , , (423)
in cui la fugacit del composto in fase liquida pu essere calcolata come discusso in
precedenza.
81
Si noti che alla pressione assegnata le condizioni di equilibrio tra la fase liquida e quella solida si
hanno per un solo valore della temperatura, quella di fusione, che diversa dalla generica T
considerata. Questo impedisce la misura sperimentale dellentalpia di fusione nelle condizioni (T, P)
considerate in quanto in tali condizioni non possono esistere le due fasi in equilibrio e ne rende quindi
necessario il calcolo attraverso un ciclo.
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
139
Nella relazione precedente la differenza tra i calori specifici molari del liquido e del solido
risulta essere solitamente piccola e il secondo termine dellesponenziale quindi spesso
trascurabile rispetto al primo termine contenente lentalpia di fusione. La fugacit del solido
pu quindi essere approssimata dalla seguente relazione:
( ) ( )
|
|
.
|
\
|
|
|
.
|
\
|
T T R
h
P T f P T f
f
fus
L S
1 1
exp , ,
(424)
4.7.4 Applicazione
Si calcoli la tensione di vapore del n-ottano a 427.9 [K] con lequazione di stato RKS.
In condizioni di equilibrio la fugacit delle due fasi deve essere uguale e quindi, dalla
relazione (401):
( ) ( ) ( ) ( ) T P T T P T
L V
= , , (425)
I coefficienti di fugacit delle due fasi possono essere calcolati, in funzione della pressione,
utilizzando unequazione di stato cubica con le relazioni riportate in Tabella 12. In ogni
caso, la relazione precedente rappresenta una equazione nella sola incognita P, che pu
quindi essere risolta.
La procedura logica per la risoluzione la seguente:
si definisce lEoS da utilizzare;
si assegna la temperatura a cui si vuole calcolare la tensione di vapore;
si assegna un valore di primo tentativo di P;
si calcolano le costanti A e B;
si risolve la equazione cubica in Z alla temperatura e pressione assegnata e si trovano i
tre valori di Z;
utilizzando il valore di Z minore si calcola (con la relazione opportuna tra quelle di
Tabella 12) il coefficiente di fugacit del liquido;
utilizzando il valore di Z maggiore si calcola (con la stessa relazione) il coefficiente di
fugacit del vapore;
se i valori calcolati dei coefficienti di fugacit del liquido e del vapore sono uguali il
valore ipotizzato della tensione di vapore corretto, altrimenti bisogna ipotizzare un
nuovo valore di P e ripetere la procedura dal punto 4.
In particolare, per lEoS RKS lequazione algebrica da risolvere la seguente:
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
140
( )
( ) 0 ln ln 1
ln ln 1
=
|
|
.
|
\
|
|
|
.
|
\
|
+
+
|
|
.
|
\
|
+
B Z
Z
B Z
B
A
Z
B Z
Z
B Z
B
A
Z
V
V
V
V
L
L
L
L
(426)
Ovviamente, la procedura illustrata in precedenza di tipo logico. La risoluzione
dellequazione algebrica non lineare pu essere effettuata con un opportuno algoritmo,
come quello illustrato di seguito in linguaggio MATLAB.
Il programma commentato e quindi di facile lettura e pu essere implementato in un file
testo di nome Pv.m per essere eseguito in ambiente MATLAB. Il programma EoS richiamato
lo stesso utilizzato nellApplicazione 3.4, mentre il calcolo della funzione da azzerare,
attraverso la funzione di MATLAB fsolve, demandato alla routine esterna phi, il cui
listato riportato di seguito alla routine Pv e pu essere implementato in un file testo di
nome phi.m.
%Pv calcolo della tensione di vapore
% usando una EoS cubica
%
% R. Rota (2002)
format compact %scrittura a video compatta
clear all %cancella tutte le variabili
close %chiude le finestre grafiche
global T Tc Pc om tipo
%dati per n-ottano
Tc =569.4; %T critica [K]
Pc =24.97e5; %P critica [Pa]
om =0.398; %fattore acentrico di Pitzer
Tn =398.9; %Teb normale [K]
tipo = 3; %EoS RKS
R = 8.314; %[J/(mol K)]
T =427.9; %Temperatura [K]
%stima del valore di P con una approssimante
%della Clapeyron basata sulla Tcritica e Teb normale
BCl=(log(Pc/1.01325E5))/(1/Tn-1/Tc); %costante A
ACl=log(1.01325E5)+BCl/Tn; %costante B
PCl=exp(ACl-BCl/T); %stima di P con Clapeyron [Pa]
P0 = fsolve('phi',PCl) %calcolo P [Pa]
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
141
function F=phi(P)
%Fi calcola il residuo dell'equazione
% F = log(Fi_L)/log(Fi_V) - 1
% dove Fi il coeff. di fugacit
% di un liquido o di un vapore puro
% usata per il calcolo della pressione di saturazione
% imponendo l'uguaglianza dei coefficienti di fugacit
%
% R. Rota (2002)
global T Tc Pc om tipo
%calcolo di Z, A e B
[Z,A,B,S,k] = EoS(T,P,Tc,Pc,om,tipo);
ZV = Z(3);
ZL = Z(1);
%calcolo coeff. di fugacit liquido e vapore con RKS
logphiL = ZL-1-log(ZL-B)-A/B*log((ZL+B)/ZL);
logphiV = ZV-1-log(ZV-B)-A/B*log((ZV+B)/ZV);
F = logphiL/logphiV - 1;
Il risolutore di equazioni algebriche non lineari fsolve richiede una stima iniziale della
soluzione. Questa stata ottenuta utilizzando la relazione di Clapeyron, le cui due costanti
sono state stimate per il n-ottano dai valori della temperatura di ebollizione a due valori di
pressione: quello atmosferico (a cui corrisponde la temperatura di ebollizione normale) e
quello critico (a cui corrisponde la temperatura critica).
Il valore calcolato con questo programma pari a 2,09 [bar], in buon accordo col valore
calcolato utilizzando lequazione di Antoine (per il n-ottano: A=15,9426; B=3120,29;
C=63,63. La relazione scritta nella forma ( ) C T B A P + = ) ln( [torr], con T in [K]),
pari a 2,13 [bar].
Un confronto tra le previsioni dellEoS RKS e quelle dellequazione di Antoine pu essere
effettuato implementando una piccola modifica al programma Pv.m come riportato di
seguito.
%PvT calcolo della tensione di vapore
% a diverse T usando una EoS cubica
%
% R. Rota (2002)
format compact %scrittura a video compatta
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
142
clear all %cancella tutte le variabili
close %chiude le finestre grafiche
global T Tc Pc om tipo
%dati per n-ottano
Tc =569.4; %T critica [K]
Pc =24.97e5; %P critica [Pa]
om =0.398; %fattore acentrico di Pitzer
Tn =398.9; %Teb normale [K]
tipo = 3; %EoS RKS
R = 8.314; %cost. gas [J/(mol K)]
TT =[300:10:400]; %vettore di T [K]
%ciclo sulla temperatura
for j=1:length(TT),
T =TT(j); %j-esima temperatura [K]
%stima del valore di P con una approssimante
%della Clapeyron basata sulla Tcritica e Teb normale
BCl =(log(Pc/1.01325E5))/(1/Tn-1/Tc); %costante A
ACl =log(1.01325E5)+BCl/Tn; %costante B
PCl =exp(ACl-BCl/T); %stima di P con Clapeyron [Pa]
P0 =fsolve('phi',PCl); %calcolo P [Pa] con RKS
PRKS(j)=P0/1e5; %memorizzo il valore
%calcolo P con equazione di Antoine [Pa]
P0 =exp(15.9426-3120.29/(T-63.63))/760*101325;
PAnt(j)=P0/1e5; %memorizzo il valore
end
semilogy(1./TT,PRKS,'-',1./TT,PAnt,'o')
axis([2.4e-3 3.4e-3 0.01 2])
xlabel('1/T [K^{-1}]')
ylabel('P [bar]')
I risultati di questi conti sono riportati nella Figura 23 su di un grafico tipo Clapeyron. Si
pu notare la buona linearit dellandamento, cos come previsto dalla relazione di
Clapeyron, e il buon accordo tra i valori previsti dallEoS RKS e quelli calcolati con la
relazione di Antoine.
4.8 Esercizi
Calcolare lentalpia e lentropia residua delletilene a 339,7 [K] e 30,7 [bar] usando
lequazione degli stati corrispondenti a tre parametri.
Risultato: -1164 [J mol
-1
];- 2,47 [J mol
-1
K
-1
].
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
143
Figura 23: confronto tra la tensione di vapore predetta con lEoS RKS (curva) e i valori
calcolati con lequazione di Antoine (simboli) per il n-ottano.
Calcolare lentalpia e lentropia residua delletilene a 339,7 [K] e 1 [bar] usando
lequazione di stato del viriale.
Risultato: -35 [J mol
-1
]; -0,07 [J mol
-1
K
-1
].
Dellammoniaca viene compressa isotermicamente e reversibilmente da 1 [bar] e 100 [C]
fino a 50 [bar.]. Si calcoli il lavoro di compressione per mole di ammoniaca utilizzando
lequazione degli stati corrispondenti a tre parametri.
Risultato: 12 [kJ mol
-1
].
Un compressore deve portare dellammoniaca da 298 [K] e 1 [bar] a 4 [bar]. Si calcoli la
temperatura della corrente uscente dal compressore e il lavoro di compressione per kg di
ammoniaca utilizzando lequazione di stato del viriale.
Risultato: 407 [K]; 235 [kJ kg
-1
].
2.4 2.5 2.6 2.7 2.8 2.9 3 3.1 3.2 3.3 3.4
x 10
-3
10
-2
10
-1
10
0
1/T [K
-1
]
P
[
b
a
r
]
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
144
Si calcoli il coefficiente di fugacit del n-butano a 50 [C] e 20 [bar] utilizzando
lequazione di stato del viriale e sapendo che la tensione di vapore del n-butano a 50 [C]
vale 4,7 [bar]. Si trascuri la correzione di Poynting.
Risultato: 0,21 [-] .
Si scarica una bombola ben coibentata di azoto di 0,15 [m
3
] con una portata costante di 10
[mol min
-1
]. Inizialmente il gas si trova a 100 [atm] e 170 [K]. Calcolare la pressione e la
temperatura del gas dopo 50 [min] assumendo che lo scarico avvenga in modo reversibile e
utilizzando le equazione di stato VdW e PR.
Risultato: 145 [K] e 49 [bar]; 144 [K] e 49 [bar].
Calcolare lentalpia e lentropia specifica del n-ottano in fase vapore a 427,9 [K] e 2,15
[bar] utilizzando come stato di riferimento il liquido saturo a 0 [C]. La tensione di vapore
del n-ottano a 0 [C] vale 0,74 [kPa]. Si utilizzi lequazione di stato PR.
Risultato: 76 [kJ mol
-1
]; 0,21 [kJ mol
-1
K
-1
].
Una persona dal peso di 60 [kg] vuole pattinare sul ghiaccio che si trova a -2 [C]. Se larea
di contatto tra la lama del pattino e il ghiaccio di 15 [mm
2
] verificare se si forma un velo
dacqua lubrificante tre la lama del pattino e il ghiaccio. Il volume specifico del ghiaccio e
dellacqua in queste condizioni rispettivamente pari a 1,091 [dm
3
kg
-1
] e 1,000 [dm
3
kg
-1
],
mentre lentalpia di fusione del ghiaccio pari a 6,002 [kJ mol
-1
].
Risultato: s.
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
145
5 Propriet delle miscele
Nei capitoli precedenti sono state discusse le propriet dei fluidi puri e si sono introdotti
diversi metodi per calcolarne i valori sulla base di poche informazioni sperimentali, quali le
grandezze critiche.
Daltro canto, nella pratica quotidiana lingegnere chimico deve svolgere dei calcoli che
coinvolgono non solo fluidi puri ma anche miscele di composti. In un certo senso, questa
una delle caratteristiche che differenzia lingegnere chimico dagli altri ingegneri, i quali
solitamente trattano fluidi puri o poche miscele di composizione ben definita. Per
definizione invece lingegnere chimico si occupa delle trasformazioni della materia, siano
esse fisiche (per esempio la separazione dei componenti una miscela, come avviene nella
distillazione del petrolio per ottenere la benzina) oppure chimiche (come avviene nelle
reazioni di polimerizzazione per produrre le materie plastiche a partire da composti puri). In
ogni caso, sono coinvolte delle miscele
82
e di conseguenza lingegnere chimico deve essere
in grado di eseguire dei calcoli che richiedono la conoscenza delle propriet delle miscele.
Inoltre, mentre dispendioso ma possibile caratterizzare sperimentalmente il
comportamento volumetrico di tutti i fluidi puri, unimpresa inimmaginabile
caratterizzare sperimentalmente il comportamento volumetrico di tutte le possibili miscele
che si possono originare dalla combinazione di tutti i composti puri in qualsiasi numero e
rapporto ponderale. Risulta quindi indispensabile sviluppare dei metodi in grado di predire
il comportamento delle miscele sulla base di poche informazioni sperimentali.
Da un punto di vista generale, come discusso in precedenza, la principale differenza tra un
fluido puro e una miscela risiede nel fatto che mentre le propriet di un sistema monofase
contenente un composto puro dipendono solo da due variabili intensive e dal numero di
moli complessivo, nel caso di miscele necessario conoscere il numero di moli di tutti i
componenti la miscela. In altri termini, oltre al numero di moli totali anche necessario
conoscere la composizione della miscela, per esempio in termini di frazione molare
83
. In
questo capitolo si svilupperanno quindi delle relazioni per caratterizzare il comportamento
di miscele omogenee di composizione variabile.
Il calcolo delle propriet di una miscela segue le stesse linee tracciate per il caso dei fluidi
puri: dapprima si sviluppano delle relazioni per il calcolo delle propriet di sistemi
particolarmente semplici, quali una miscela di gas perfetti, e poi si ricercano delle
82
Nessuna separazione o reazione chimica ha infatti senso per un composto puro.
83
Si ricorda che si definisce frazione molare di un generico composto i presente in una miscela il
rapporto tra il numero di moli del composto i e il numero di moli totali della miscela, x
i
=n
i
/n
tot
. Nel
caso di sistemi multifase, la frazione molare di un composto riferita alla miscela presente in una data
fase (per esempio, quella vapore) ed definita come il rapporto tra il numero di moli del composto i
presenti nella fase vapore e il numero di moli totali della miscela in fase vapore, x
i
V
=n
i
V
/n
tot
V
.
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
146
espressioni che consentano il calcolo dello scostamento del comportamento dei sistemi reali
da quello di gas perfetto, cio il calcolo delle grandezze residue. Anche nel caso di miscele,
il calcolo delle correzioni rispetto al comportamento di gas perfetto risulta essere pi
affidabile per la fase gassosa che per le fasi condensate. Analogamente al caso dei composti
puri, possibile migliorare laffidabilit delle previsioni delle propriet di fasi condensate
introducendo delle ulteriori informazioni sperimentali.
Al termine dello studio di questo capitolo lo studente dovrebbe conoscere:
la definizione e il significato di grandezza parziale molare;
la relazione tra le grandezze parziali molari e le grandezze di miscela;
la relazione di Gibbs Duhem;
il teorema di Gibbs per le miscele di gas perfetti;
come calcolare le propriet di una miscela di gas perfetti;
la definizione di propriet di miscelazione;
come calcolare le propriet di miscelazione per una miscela di gas perfetti;
cosa sono le regole di miscelazione e di combinazione;
le regole di miscelazione per equazioni di stato cubiche, del viriale e per la legge degli
stati corrispondenti;
come calcolare il coefficiente di compressibilit e le funzioni residue di una miscela
con diverse EoS;
la definizione di fugacit e coefficiente di fugacit di un composto in una miscela;
come calcolare la fugacit di un composto in miscela con una EoS;
cosa una miscela ideale;
come calcolare le propriet parziali molari di una miscela ideale;
come calcolare le propriet di una miscela ideale;
come calcolare le propriet di miscelazione per una miscela ideale;
la regola di Lewis - Randall;
la definizione e il significato di funzione di eccesso;
la definizione di coefficiente di attivit e la sua relazione con lenergia libera di Gibbs
molare di eccesso;
la relazione tra le altre grandezze termodinamiche e lenergia libera di Gibbs molare di
eccesso;
come si misurano sperimentalmente i coefficienti di attivit;
i modelli di Margules, van Laar, Wilson.
5.1 Propriet delle miscele
La relazione fondamentale per la descrizione delle propriet di una miscela la (237)
=
+ + =
N
i
i i
dn VdP SdT dG
1
(427)
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
147
che mostra come lenergia libera di Gibbs di una miscela dipenda da T, P e n: G =
G(T,P,n). Ne consegue che, essendo:
|
|
.
|
\
|
+ |
.
|
\
|
+ |
.
|
\
|
=
N
i
n P T
i T P
i j
n
G
dP
P
G
dT
T
G
dG
1
, ,
, , n n
(428)
Devono valere le seguenti uguaglianze:
i j
n P T
i
i
T
P
n
G
P
G
V
T
G
S
|
|
.
|
\
|
=
|
.
|
\
|
=
|
.
|
\
|
=
, ,
,
,
n
n
(429)
Queste relazioni possono anche essere scritte per il caso particolare di una mole di miscela,
per cui le grandezze estensive coincidono con le relative grandezze molari e il numero di
moli di ciascuna specie con le corrispondenti frazioni molari:
=
+ + =
N
i
i i
dx vdP sdT dg
1
(430)
che mostra come g = g(T,P,x). Poich per le frazioni molari non sono variabili
indipendenti (in quanto 1
1
=
=
N
i
i
x ), si possono stabilire solo le seguenti due relazioni:
n
n
,
,
T
P
P
g
v
T
g
s
|
.
|
\
|
=
|
.
|
\
|
=
(431)
Si nota come anche nel caso delle soluzioni g pu essere vista come la funzione generatrice
di tutte le altre grandezze termodinamiche, in quanto da essa si possono calcolare s e v con
operazioni di derivazione, e di conseguenza tutte le altre propriet con operazioni di somma
e moltiplicazione: h = g + Ts, u = h - Pv, ecc.
Inoltre, la relazione (240) pu essere vista come un caso particolare della (430) per un
sistema a composizione costante.
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
148
5.1.1 Propriet parziali molari
Data una generica propriet di miscela M = M(T,P,n), la variazione di tale propriet
dipende dalla variazione di T, P e n:
=
|
|
.
|
\
|
+ |
.
|
\
|
+ |
.
|
\
|
=
N
i
i
n P T
i T P
dn
n
M
dP
P
M
dT
T
M
dM
i j
1
, ,
, , n n
(432)
Per il ruolo assolutamente centrale che gioca nella termodinamica delle miscele, si assegna
un nome particolare allultima derivata parziale definendola grandezza parziale molare,
indicata con
i
M :
i j
n P T
i
i
n
M
M
|
|
.
|
\
|
=
, ,
(433)
Alla grandezza parziale molare pu essere associato il significato fisico di valore della
grandezza molare del composto i quando questo in una certa miscela
84
.La relazione (432)
pu essere riscritta in termini di grandezze molari come:
( ) ( )
=
|
|
.
|
\
|
+ |
.
|
\
|
+ |
.
|
\
|
=
N
i
i
n P T
i T P
dn
n
M
dP
P
nm
dT
T
nm
nm d
i j
1
, ,
, ,
) (
n n
(434)
Ricordando che le prime due derivate parziali sono effettuate a numero di moli totali
costante, si ottiene:
( )
=
+ + |
.
|
\
|
+ |
.
|
\
|
= +
N
i
i i i
T P
dn x ndx M dP
P
m
n dT
T
m
n mdn ndm
1 , , n n
(435)
in cui si anche utilizzata la definizione di frazione molare n
i
= n x
i
, da cui si ottiene dn
i
=
nd x
i
,+ x
i
dn.
Riarrangiando la relazione precedente si ottiene:
0
1 1 , ,
=
|
|
.
|
\
|
+
|
|
.
|
\
|
|
.
|
\
|
|
.
|
\
|
= =
N
i
i i
N
i
i i
T P
x M m dn dx M dP
P
m
dT
T
m
dm n
n n
(436)
84
Questo significa che la grandezza molare di un composto pu assumere valori diversi se il
composto puro o in miscela con altri composti. Per esempio, una mole di un composto pu occupare
un volume differente se il composto puro o in miscela. Questa interpretazione corretta ricordando
che la definizione di grandezza parziale molare assegna arbitrariamente a ciascun composto presente
nella miscela una porzione della propriet M. Altre assegnazioni, egualmente valide, sono possibili.
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
149
Poich n e dn sono indipendenti, luguaglianza precedente deve essere vera per qualsiasi
valore di n e dn. Questo possibile solo se i due termini tra parentesi sono nulli. Il primo:
=
+ |
.
|
\
|
+ |
.
|
\
|
=
N
i
i i
T P
dx M dP
P
m
dT
T
m
dm
1 , , n n
(437)
non fornisce informazioni di particolare rilevanza in quanto si tratta di un caso particolare
della relazione (432) riferita a una mole di miscela. Ben pi importante la seconda
relazione:
=
=
N
i
i i
x M m
1
(438)
che definisce la grandezza molare di miscela come una media pesata sulle frazioni molari
delle relative grandezze parziali molari. Moltiplicando ambo i membri per il numero di
moli totali si ottiene la seguente relazione:
=
=
N
i
i i
n M M
1
(439)
Queste relazioni consentono il calcolo delle propriet di miscela sulla base del valore delle
grandezze parziali molari e della composizione. Il problema del calcolo delle propriet di
una miscela si riduce quindi al calcolo delle relative propriet parziali molari
85
.
Tutte le propriet parziali molari si possono ricavare dalla energia libera di Gibbs (che al
solito pu essere considerata come la funzione generatrice delle altre) attraverso le relazioni
dedotte dalle equazioni (429):
i i i
S T G H + = (440)
i i i
V P H U = (441)
85
Un esempio facilmente intuibile riguarda il volume. Miscelando un volume V
A
= n
A
v
A
del
composto A con un volume V
B
= n
B
v
B
del composto B, il volume della miscela risultante sar pari a
B B A A
V n V n V + = . Solo nel caso particolare in cui i volumi molari dei composti puri coincidono
coi rispettivi volumi parziali molari il volume della soluzione risulta uguale alla somma dei volumi
dei composti puri miscelati. In altri termini, miscelando 1 litro del composto A con 1 litro del
composto B non sempre si ottengono 2 litri di miscela.
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
150
x
x
n
,
,
, ,
, ,
,
, ,
P
i
P
n P T
i
n P T
P i
n P T
i
i
T
G
n
G
T
T
G
n n
S
S
i j
i j
i j
|
|
.
|
\
|
=
|
|
|
.
|
\
|
|
|
.
|
\
|
=
=
|
|
.
|
\
|
|
.
|
\
|
=
|
|
.
|
\
|
(442)
x
x
n
,
,
, ,
, ,
,
, ,
T
i
T
n P T
i
n P T
T i
n P T
i
i
P
G
n
G
P
P
G
n n
V
V
i j
i j
i j
|
|
.
|
\
|
=
|
|
|
.
|
\
|
|
|
.
|
\
|
=
=
|
|
.
|
\
|
|
.
|
\
|
=
|
|
.
|
\
|
(443)
5.1.1.1 Applicazione
Volendo preparare 3 [m
3
] di una miscela etanolo / acqua al 60% in moli di etanolo, si
calcoli il volume di acqua e di metanolo da miscelare. I volumi molari di acqua e metanolo
sono pari a 57,9 10
-6
e 18 10
-6
[m
3
mol
-1
] rispettivamente, mentre i volumi parziali molari
della miscela da preparare sono pari a 57,5 10
-6
e 16 10
-6
[m
3
mol
-1
] rispettivamente.
Il volume molare della soluzione da preparare dato da:
] mol [m 10 9 , 40 4 , 0 10 16 6 , 0 10 5 , 57
1 3 6 6 6
1
=
= + = =
N
i
i i
x V v (444)
Le moli di soluzione da preparare sono quindi:
[mol] 6 , 73349
10 9 , 40
3
6
=
= =
v
V
n
(445)
di cui
[mol] 8 , 44009 6 , 0 6 , 73349
1 1
= = = x n n (446)
di etanolo (composto 1), e
[mol] 8 , 29339 4 , 0 6 , 73349
2 2
= = = x n n (447)
di acqua (composto 2).
Il volume di etanolo da miscelare quindi pari a:
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
151
] [m 548 , 2 10 9 , 57 8 , 44009
3 6
1 1 1
= = =
v n V (448)
mentre quello di acqua pari a:
] [m 528 , 0 10 18 8 , 29339
3 6
2 2 2
= = =
v n V (449)
Si noti che miscelando 2,548+0,528=3,076 [m
3
] di composti puri si ottengono 3 [m
3
] di
miscela.
5.1.2 Equazione di Gibbs - Duhem
86
Unaltra importante relazione pu essere ricavata dalle due relazioni derivate nella sezione
precedente, (437) e (438). Differenziando la relazione (438) e inserendo la (437) si ottiene:
= = =
+ |
.
|
\
|
+ |
.
|
\
|
= + =
N
i
i i
T P
N
i
i i
N
i
i i
dx M dP
P
m
dT
T
m
M d x dx M dm
1 , , 1 1 n n
(450)
da cui si deduce la cosiddetta equazione di Gibbs Duhem:
0
1 , ,
= |
.
|
\
|
+ |
.
|
\
|
=
N
i
i i
T P
M d x dP
P
m
dT
T
m
n n
(451)
che deve essere soddisfatta per tutte le variazioni di temperatura, pressione e composizione
in una miscela omogenea. A temperatura e pressione costante questa relazione assume la
forma pi nota:
0
1
=
=
N
i
i i
M d x (452)
5.2 Miscele di gas perfetti
Le grandezze parziali molari di una miscela di gas perfetti si possono calcolare sulla base
del cosiddetto teorema di Gibbs:
Tutte le propriet parziali molari, eccetto il volume, di un componente una miscela di gas
perfetti sono uguali alle corrispondenti propriet molari del composto puro come gas
perfetto calcolate alla stessa temperatura della miscela ma a una pressione uguale alla
pressione parziale
87
del componente:
86
Pierre Maurice Marie Duhem, 1861 1916, fisico francese.
87
Si ricorda che la pressione parziale di un composto definita come la pressione del sistema
moltiplicata per la frazione molare del composto: P
i
= P x
i
.
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
152
) , ( ) , , (
* *
i i i
P T m P T M = x (453)
Questo non vale ovviamente per il volume, in quanto:
( )
) , (
) , , (
*
, ,
1
, , , ,
*
*
P T v
P
RT
n
n
P
RT
n
P nRT
n
V
P T V
i
n P T
i
N
k
k
n P T
i
n P T
i
i
i j
i j i j
= =
|
|
|
|
.
|
\
|
|
.
|
\
|
=
=
|
|
.
|
\
|
=
|
|
.
|
\
|
=
x
(454)
dove si sfruttato il fatto che il numero di moli totale dato dalla somma del numero di
moli di tutti i componenti la miscela e che la derivata parziale viene effettuata mantenendo
costante il numero di moli di tutti i composti tranne quello rispetto a cui si effettua la
derivazione.
Lentalpia parziale molare si calcola quindi come:
) ( ) , ( ) , , (
* * *
T h P T h P T H
i i i i
= = x (455)
in quanto per un gas perfetto lentalpia molare non dipende dalla pressione. Lentalpia
molare di una miscela di gas perfetti si calcola quindi semplicemente come media pesata
sulla composizione dei valori delle entalpie molari dei composti puri alla stessa temperatura
del sistema:
= =
= =
N
i
i i
N
i
i i
x T h x P T H P T h
1
*
1
* *
) ( ) , , ( ) , , ( x x (456)
Un discorso analogo vale per lenergia interna:
= =
= =
N
i
i i
N
i
i i
x T u x P T U P T u
1
*
1
* *
) ( ) , , ( ) , , ( x x (457)
Dalle due ultime relazioni insieme alle definizioni di calore specifico si ricavano facilmente
anche le relazioni per il calcolo dei calori specifici per una miscela di gas perfetti:
=
=
N
i
i i P P
x T c P T c
1
*
,
*
) ( ) , , ( x (458)
=
=
N
i
i i V V
x T c P T c
1
*
,
*
) ( ) , , ( x (459)
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
153
Risulta evidente lestrema semplicit del calcolo delle propriet di una miscela di gas
perfetti, legato essenzialmente al fatto che la dipendenza dalla composizione risulta essere
lineare, cio la pi semplice possibile.
Appena un po pi complicato il calcolo delle propriet che dipendono anche dalla
pressione, come lentropia:
) , ( ) , , (
* *
i i i
P T s P T S = x (460)
Ricordando che per un gas perfetto:
P Rd ds P Rd dT
T
c
ds
T
P
ln ln
*
= =
(461)
si ottiene che
i
i i
P
P
i
P
P
T
x R
Px
P
R
P
P
R P Rd P T s P T s ds
i i
ln ln ln ln ) , ( ) , ( =
|
|
.
|
\
|
=
|
|
.
|
\
|
= = =
(462)
Inserendo questa equazione nella (460) si ottiene:
i i i i i
x R P T s P T s P T S ln ) , ( ) , ( ) , , (
* * *
= = x (463)
e quindi lentropia molare della miscela calcolata come:
= = =
= =
N
i
i i
N
i
i i
N
i
i i
x x R x P T s x P T S P T s
1 1
*
1
* *
ln ) , ( ) , , ( ) , , ( x x (464)
Analogamente, lenergia libera di Gibbs della miscela si calcola come:
i i i i i
i i i i i i
x RT P T g x RT P T Ts T h
P T Ts T h P T g P T G
ln ) , ( ln ) , ( ) (
) , ( ) ( ) , ( ) , , (
* * *
* * * *
+ = + =
= = = x
(465)
= = =
+ = =
N
i
i i
N
i
i i
N
i
i i
x x RT x P T g x P T G P T g
1 1
*
1
* *
ln ) , ( ) , , ( ) , , ( x x (466)
Ricordando che il potenziale chimico di un composto in miscela coincide con lenergia
libera di Gibbs parziale molare, mentre quello di un composto puro con lenergia libera di
Gibbs molare, si ottiene dalla (465):
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
154
|
|
.
|
\
|
+ =
|
|
.
|
\
|
+ =
= +
|
|
.
|
\
|
+ =
= + =
r
i
r i
r
i
r i
i
r
r i
i i i
P
P
RT P T
P
Px
RT P T
x RT
P
P
RT P T
x RT P T P T
ln ) , ( ln ) , (
ln ln ) , (
ln ) , ( ) , , (
* *
*
* *
x
(467)
dove stata utilizzata anche la relazione (394) inserendo un valore di riferimento per la
pressione, P
r
. Differenziando questa espressione si ottiene una relazione tra potenziale
chimico e pressione parziale analoga a quella valida per i gas perfetti puri:
i i
P RTd P T d ln ) , , (
*
= x (468)
5.2.1 Propriet di miscelazione
Come discusso in precedenza, quando dei fluidi puri vengono miscelati, le propriet della
miscela che si forma possono essere diverse dalla somma delle propriet dei fluidi puri di
partenza. Si pu quindi avere una variazione delle propriet a causa della miscelazione.
Queste variazioni vanno sotto il nome di propriet di miscelazione, definite come
differenza tra la propriet della miscela e la somma delle propriet dei fluidi puri che,
miscelati, formano la miscela in esame:
= =
=
=
= =
N
i
i i
N
i
i i
N
i
i i
mix
P T m n P T M n
P T m n P T M P T M
1 1
1
) , ( ) , (
) , ( ) , , ( ) , , ( x x
(469)
Dalla relazione precedente si ricava la definizione di propriet di miscelazione molare
dividendo tutti i membri per il numero di moli totali della miscela:
= =
=
=
=
N
i
i i
N
i
i i
N
i
i i
mix
P T m x P T M x
P T m x P T m P T m
1 1
1
) , ( ) , (
) , ( ) , , ( ) , , ( x x
(470)
Dalle reazioni ricavate in precedenza per le diverse propriet di una miscela di gas perfetti
si pu ricavare che il volume, lentalpia e lenergia interna molare di miscelazione sono
nulli, come logico attendersi poich in un gas perfetto tutte le interazioni steriche ed
energetiche tra le molecole sono nulle:
0 ) ( ) , ( ) , , (
1
* * *,
= =
=
N
i
i i
mix
T h x T h P T h x x (471)
0 ) ( ) , ( ) , , (
1
* * *,
= =
=
N
i
i i
mix
T u x T u P T u x x (472)
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
155
0 ) , ( ) , , ( ) , , (
1 1
* * *,
= =
= = =
N
i
i
N
i
i i
mix
P
RT
x
P
RT
P T v x P T v P T v x x (473)
Il fatto che il volume di miscelazione sia nullo significa che miscelando 1 litro del
composto A con 1 litro del composto B si ottengono 2 litri di miscela. Il processo di
miscelazione non cio associato a una variazione del volume.
Il fatto che lentalpia (o lenergia interna) di miscelazione sia nulla significa che miscelando
senza scambiare energia con lambiente a pressione (o volume) costante il composto A a 20
[C] con il composto B a 20 [C] si ottiene una miscela a 20 [C]. Il processo di
miscelazione cio atermico.
Viceversa, lentropia molare di miscelazione diversa da zero e pari a:
0 ln
) , ( ln ) , (
) , ( ) , , ( ) , , (
1
1
*
1 1
*
1
* * *,
> =
= =
= =
=
=
= =
=
N
i
i i
N
i
i i
N
i
i i
N
i
i i
N
i
i i
mix
x x R
P T s x x x R P T s x
P T s x P T s P T s x x
(474)
Questo mostra come alla miscelazione di gas perfetti associato un aumento di entropia.
Infatti poich x
i
< 1, deve essere ln x
i
< 0 e quindi lentropia di miscelazione risulta sempre
positiva. In altri termini, la miscelazione di gas perfetti un processo spontaneo.
5.2.2 Applicazione
Una bombola da 60 [l] viene riempita di una miscela di azoto e anidride carbonica fino alla
pressione di 40 [bar] collegandola con una linea a 50 [bar] e 20 [C]. La pressurizzazione
della bombola cos rapida che la si pu assumere adiabatica. Se la bombola contiene
inizialmente la miscela a 1 [bar] e 20 [C], quale sar la temperatura alla fine della carica di
una miscela contenente il 10% o il 90% di azoto? Si consideri la miscela un gas perfetto. I
calori specifici a volume costante dellazoto e della CO
2
come gas perfetto si assumano
costanti e pari rispettivamente a 21 e 33 [J mol
-1
K
-1
].
Questo problema gi stato affrontato nellApplicazione 2.6 per il caso di un fluido
assimilabile a un fluido puro e quindi si riporta di seguito solo la relazione risolutiva:
|
|
.
|
\
|
+
=
IN
P
V
T
P P
c
c
T
P
P
T
1 2
*
*
1
1
2
2
(475)
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
156
Il calore specifico a volume costante della miscela si calcola, per i due casi considerati,
come:
= +
= +
= =
=
] K mol [J 2 , 22 33 1 , 0 21 9 , 0
] K mol [J 8 , 31 33 9 , 0 21 1 , 0
) ( ) , , (
1 1
1 1
1
*
,
*
N
i
i i V V
x T c P T c x (476)
La temperatura finale nei due casi risulta quindi pari a:
=
|
.
|
\
|
+
+
=
|
.
|
\
|
+
+
=
[K] 367
293
1 40
314 , 8 8 , 31
8 , 31
293
1
40
[K] 399
293
1 40
314 , 8 2 , 22
2 , 22
293
1
40
2
T (477)
5.3 Funzioni residue da EoS
Laria una miscela a composizione approssimativamente costante costituita
essenzialmente da azoto e ossigeno, ma le sue propriet termodinamiche sono solitamente
calcolate come se si trattasse di un fluido puro. Questo mostra intuitivamente come
dovrebbe essere possibile modificare le equazioni di stato trattate in precedenza per
prevedere il comportamento volumetrico anche delle miscele.
In particolare, ci si pu attendere che le stesse relazioni matematiche discusse in precedenza
siano in grado di rappresentare anche il comportamento volumetrico delle miscele pur di
sostituire ai parametri relativi ai composti puri quelli relativi alla miscela in esame. Le
equazioni che esprimono i parametri delle diverse equazioni di stato in funzione della
composizione della miscela e dei parametri dei composti puri sono chiamate regole di
miscelazione. Queste regole di miscelazione sono essenzialmente empiriche e devono la
loro validit alla loro capacit di prevedere il comportamento sperimentale
88
delle miscele
in differenti condizioni.
In generale le regole di miscelazione cercano di predire i parametri di una generica miscela
sulla base di parametri relativi a coppie di composti. Da un lato questo approccio ha una
giustificazione teorica basata sul fatto di ritenere che le interazioni tra due molecole siano
pi importanti che non quelle tra tre o pi molecole, ma dallaltra ha un enorme rilevanza
dal punto di vista applicativo. Poich i parametri coinvolti sono relativi solo a coppie di
composti, possibile stimare tutti i parametri necessari per prevedere il comportamento di
88
Fanno eccezione la regola di miscelazione per il secondo coefficiente del viriale, che pu essere
dedotta in modo esatto dalla meccanica statistica, e le regole da essa derivate, come quelle di Wong-
Sandler (D.S.H. Wong, S.I. Sandler, AIChE J., 38, 671, 1992).
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
157
una miscela di N composti per confronto coi dati sperimentali di tutte e sole le possibili
miscele binarie che si possono formare dagli N componenti la miscela. I parametri binari
stimati dai dati sperimentali relativi a una data miscela bicomponente saranno sempre gli
stessi indipendentemente dagli altri composti presenti nella miscela a N componenti.
Questo riduce enormemente il numero di informazioni sperimentali che devono essere
raccolte.
La forma pi semplice delle regole di miscelazione in grado di fornire risultati ragionevoli
per un parametro generico m la seguente, detta quadratica o di van der Waals:
Mx x
T
= =
= =
N
i
N
j
ij j i
m x x m
1 1
(478)
dove x
i
e x
j
sono le frazioni molari della specie i e j, mentre m
ii
e m
jj
sono i parametri
relativi ai composti i e j puri. Viceversa, m
ij
sono i parametri di interazione binaria tra i
composti i e j. M rappresenta la matrice con elementi m
ij
, mentre x il vettore riga con
elementi x
i
e lapice
T
indica il vettore trasposto
89
.
Le equazioni che forniscono i parametri di interazione binaria in funzione dei parametri dei
composti puri sono chiamate regole di combinazione. Le due regole di combinazione pi
semplici sono la media aritmetica (solitamente utilizzata per parametri geometrici):
2
j i
ij
m m
m
+
= (479)
per cui la regola di miscelazione (478) si riduce alla:
=
=
N
i
i i
m x m
1
(480)
e la media geometrica (solitamente utilizzata per parametri energetici):
j i ij
m m m = (481)
per cui la regola di miscelazione (478) si riduce alla:
2
1
|
.
|
\
|
=
=
N
i
i i
m x m (482)
Una volta definite le regole di miscelazione per una delle EoS discusse in precedenza,
quindi possibile calcolare il coefficiente di compressibilit e le funzioni residue (entalpia,
entropia, energia interna, energia libera di Gibbs e di Helmholtz) per una miscela. Da
queste informazioni, poi ovviamente possibile, analogamente a quanto visto in
precedenza, calcolare il volume molare e il valore delle funzioni di interesse per la miscela.
89
La notazione vettoriale risulta particolarmente conveniente per limplementazione in linguaggi
evoluti, quale MATLAB.
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
158
5.3.1 Equazioni di stato cubiche
Le regole di combinazione pi semplici per i parametri a e b presenti nelle equazioni di
stato cubiche sono le seguenti:
( )
ij j i ij
k a a a = 1 (483)
2
j i
ij
b b
b
+
=
(484)
coerentemente col fatto che b un parametro geometrico e a energetico. Il parametro k
ij
rappresenta le interazioni tra le molecole di tipo i e quelle di tipo j e deve essere stimato da
dati sperimentali relative a miscele i-j. In assenza di informazioni, pu essere posto uguale
a zero
90
. Si noti che in questo caso possibile prevedere il comportamento di una miscela
sulla base di informazioni relative ai soli composti puri.
Le relazioni riportate nei paragrafi precedenti per il calcolo del coefficiente di
compressibilit e delle funzioni residue dei composti puri rimangono immutate pur di
sostituire i parametri a e b coi rispettivi valori della miscela. Lunico parametro che deve
essere calcolato in maniera differente
91
il parametro contenuto nelle espressioni delle
funzioni residue delle EoS RKS e PR che, nel caso di miscele assume la forma:
a
k
T a
S x
N
i
Ri i
i i
=
=
1 1
(485)
5.3.2 Equazione del viriale
La regola di combinazione pi semplice per il secondo coefficiente del viriale la seguente:
( )
1 0
B B
P
RT
B
ij
Cij
Cij
ij
+ =
(486)
dove B
0
e B
1
vengono calcolate con le relazioni (176) alla temperatura ridotta T
R
= T/T
Cij
.
La temperatura pseudocritica viene calcolata con una media geometrica:
90
Questa approssimazione tanto pi corretta quanto pi i due composti sono chimicamente simili,
cio, come verr discusso pi avanti, tanto pi la miscela si avvicina al comportamento di una miscela
ideale.
91
Lespressione seguente pu facilmente essere dedotta seguendo la derivazione delle espressioni
delle funzioni residue discussa in precedenza.
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
159
( )
ij Cj Ci Cij
k T T T = 1 (487)
Il parametro k
ij
, che uguale a zero per i = j e per composti chimicamente simili,
rappresenta le interazioni tra le molecole di tipo i e quelle di tipo j e deve essere stimato da
dati sperimentali relative a miscele i-j. In assenza di informazioni, pu essere posto uguale
a zero oppure stimato con la relazione:
Cij
Cj Ci
Cij
v
v v
k =1 (488)
dove il volume pseudocritico viene calcolato dalla relazione:
3
3 / 1 3 / 1
2 |
|
|
.
|
\
|
+
=
Cj Ci
v v
v
Cij
(489)
Il fattore acentrico della miscela viene solitamente calcolato con una media aritmetica:
2
j i
ij
+
=
(490)
mentre la pressione pseudocritica viene calcolata dalla definizione del fattore di
compressibilit:
Cij
Cij Cij
Cij
v
RT Z
P =
(491)
in cui:
2
Cj Ci
Cij
Z Z
Z
+
= (492)
Stimando il valore del parametro k
ij
con la relazione (488) possibile prevedere il
comportamento di una miscela sulla base di informazioni relative ai soli composti puri. I
risultati risultano tanto pi accurati tanto pi i composti sono chimicamente simili.
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
160
5.3.3 Equazione degli stati corrispondenti
Le regole di combinazione pi semplici che possono essere utilizzate con lequazione degli
stati corrispondenti per il calcolo di una temperatura e pressione pseudocritica della
miscela
92
sono quelle di Kay
93
basate su una media aritmetica:
2
Cj Ci
Cij
T T
T
+
=
(493)
2
Cj Ci
Cij
P P
P
+
= (494)
per cui la regola di miscelazione si riduce alla:
=
=
N
i
Ci i C
T x T
1
(495)
=
=
N
i
Ci i C
P x P
1
(496)
e le variabili ridotte per la miscela si calcolano come:
C
R
T
T
T =
(497)
C
R
P
P
P =
(498)
La regola di combinazione per il fattore acentrico solitamente usata quella di Joffe
94
basata anchessa su una media aritmetica:
2
j i
ij
+
=
(499)
per cui la regola di miscelazione anche per il fattore acentrico si riduce alla:
=
=
N
i
i i
x
1
(500)
92
Queste regole non devono essere intese fornire la reale temperatura e pressione critica della
miscela, ma piuttosto valori efficaci in grado di prevedere valori ragionevoli delle variabili ridotte per
lutilizzo dellequazione degli stati corrispondenti.
93
W.B. Kay, Ind. Eng. Chem., 28, 1014 (1936).
94
J. Joffe, Ind. Eng. Chem. Fund., 10, 532 (1971).
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
161
La relazione di Kay per la pressione pseudocritica non risulta soddisfacente quando nella
miscela sono presenti composti chimicamente molto diversi. In questo caso si ottengono
risultati migliori con la regola di Prausnitz - Gunn
95
:
=
=
=
N
i
Ci i
N
i
Ci i C
C
v x
Z x RT
P
1
1
(501)
In tutti i casi si prevede il comportamento di una miscela sulla base di informazioni relative
ai soli composti puri e i risultati risultano tanto pi accurati tanto pi i composti sono
chimicamente simili.
5.4 Applicazione
Si calcoli il coefficiente di compressibilit e lentalpia residua di una miscela equimolare di
n-butano e n-ottano in fase vapore a 600 [K] e 16 [bar] con le diverse equazioni di stato
viste in precedenza trascurando i parametri di interazione binaria nelle regole di
miscelazione.
Gas ideale
0
1
=
=
R
h
Z
(502)
vdW
I valori dei parametri per i composti puri si calcolano come visto nei capitoli precedenti. Si
ottiene per il n-ottano:
] mol m [J 7863 , 3
2 3
= a
] [m 3698 0002 , 0
3
= b
(503)
mentre per il n-butano:
] mol m [J 1,3885
2 3
= a
] [m 0,00011638
3
= b
(504)
I parametri per la miscela si calcolano quindi come:
95
J.M. Prausnitz, R.D. Gunn, AIChE J., 4, 494 (1958).
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
162
] mol m [J 2,4402
2 3
2
1
=
= |
.
|
\
|
=
N
i
i i
a x a
] [m 0,00017668
3
1
= =
=
N
i
i i
b x b
(505)
( )
1569 , 0
2
= =
RT
aP
A
056669 , 0 = =
RT
bP
B
(506)
-1,0567 =
0,1569 =
-0,0088913 =
(507)
-0,21529 = p
-0,041023 = q
5
10 5,1158
= D
(508)
Poich D > 0 lequazione ammette una sola soluzione reale:
0,89194 = Z (509)
Utilizzando questi valori si pu calcolare il valore dellentalpia residua:
28397 , 0
89194 , 0
1569 , 0
1 89194 , 0 1 = = =
Z
A
Z
RT
h
R
(510)
da cui:
] mol [J 1416,5 6000 314 , 8 28397 , 0
1
= = = RT
RT
h
h
R
R
(511)
RK
Per il n-ottano:
] mol m [J 3,7375
2 3
= a
] [m 00016426 , 0
3
= b
(512)
Per il n-butano:
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
163
] mol m [J 1,1844
2 3
= a
] [m 4 0,00008066
3
= b
(513)
I parametri per la miscela si calcolano quindi come:
] mol m [J 2,2825
2 3
2
1
=
= |
.
|
\
|
=
N
i
i i
a x a
] [m 0,00012246
3
1
= =
=
N
i
i i
b x b
(514)
( )
14676 , 0
2
= =
RT
aP
A
039279 , 0 = =
RT
bP
B
(515)
-1,0000 =
10594 , 0 =
-0,0057645 =
(516)
-0,2274 = p
-0,044526 = q
5
10 6,0147
= D
(517)
Poich D > 0 lequazione ammette una sola soluzione reale:
0,88801 = Z (518)
Utilizzando questi valori si pu calcolare il valore dellentalpia residua:
35456 , 0 ln
2
3
1 = |
.
|
\
| +
=
Z
B Z
B
A
Z
RT
h
R
(519)
] mol [J 1768,7
1
= = RT
RT
h
h
R
R
(520)
RKS
Per il n-ottano:
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
164
] mol m [J 3,6203
2 3
= a
] [m 00016426 , 0
3
= b
(521)
Per il n-butano:
] mol m [J 1,022
2 3
= a
] [m 4 0,00008066
3
= b
(522)
I parametri per la miscela si calcolano quindi come:
] mol m [J 2,1223
2 3
2
1
=
= |
.
|
\
|
=
N
i
i i
a x a
] [m 0,00012246
3
1
= =
=
N
i
i i
b x b
(523)
( )
13646 , 0
2
= =
RT
aP
A
039279 , 0 = =
RT
bP
B
(524)
-1,0000 =
095639 , 0 =
-0,00536 =
(525)
-0,23769 = p
-0,047555 = q
5
10 6,7971
= D
(526)
Poich D > 0 lequazione ammette una sola soluzione reale:
0,90039 = Z (527)
Utilizzando questi valori si pu calcolare il valore del parametro :
1,1246
1 1
= =
=
a
k
T a
S x
N
i
Ri i
i i
(528)
e quindi quello dellentalpia residua:
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
165
( ) 41477 , 0 ln 1 1 = |
.
|
\
| +
+ =
Z
B Z
B
A
Z
RT
h
R
(529)
] mol [J 2069,0
1
= = RT
RT
h
h
R
R
(530)
PR
Per il n-ottano:
] mol m [J 3,9005
2 3
= a
] [m 0001475 , 0
3
= b
(531)
Per il n-butano:
] mol m [J 1,1494
2 3
= a
] [m 4 0,00007243
3
= b
(532)
I parametri per la miscela si calcolano quindi come:
] mol m [J 2,3212
2 3
2
1
=
= |
.
|
\
|
=
N
i
i i
a x a
] [m 0,00012246
3
1
= =
=
N
i
i i
b x b
(533)
( )
14925 , 0
2
= =
RT
aP
A
035271 , 0 = =
RT
bP
B
(534)
-0,96473 =
074972 , 0 =
-0,0039762 =
(535)
-0,23526 = p
-0,046376 = q
5
10 5,5416
= D
(536)
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
166
Poich D > 0 lequazione ammette una sola soluzione reale:
0,8851 = Z (537)
Utilizzando questi valori si pu calcolare il valore del parametro :
96623 , 0
1 1
= =
=
a
k
T a
S x
N
i
Ri i
i i
(538)
e quindi quello dellentalpia residua:
( )
( )
( )
43406 , 0
2 1
2 1
ln 1
2 2
1 =
|
|
.
|
\
|
+
+ +
+ + =
B Z
B Z
B
A
Z
RT
h
R
(539)
] mol [J 2165,2
1
= = RT
RT
h
h
R
R
(540)
Stati corrispondenti
I valori delle costanti pseudocritiche si calcolano, con la regola di Kay, come:
[K] 3 , 497
1
= =
=
N
i
Ci i C
T x T
[bar] 47 , 31
1
= =
=
N
i
Ci i C
P x P
298 , 0
1
= =
=
N
i
i i
x
(541)
Quindi le variabili ridotte sono T
R
= 1,21 e P
R
= 0,51. Dalle tabelle di Lee - Kesler a T
R
=
1,21 e P
R
= 0,51 si ricava:
Z
0
= 0,90
Z
1
= 0,03
(542)
e quindi
91 , 0
1 0
= + = Z Z Z (543)
Si pu analogamente calcolare:
42 , 0
0
=
(
(
C
R
RT
h
(544)
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
167
06 , 0
1
=
(
(
C
R
RT
h
( ) 48 , 0 06 , 0 298 , 0 42 , 0
1 0
= + =
(
(
+
(
(
=
C
R
C
R
C
R
RT
h
RT
h
RT
h
(545)
] mol [J 7 , 1991 3 , 497 314 , 8 48 , 0
1
= = =
C
C
R
R
RT
RT
h
h (546)
Viriale
I valori dei parametri binari i-j (indicando con il pedice 1il n-ottano e 2 il n-butano) si
calcolano come (si noti che, per come sono definiti, i termini ii e jj coincidono con gli
analoghi termini dei composti i e j puri):
(
=
(
(
(
(
+
+
=
274 , 0 0,265
0,265 256 , 0
2
2
2
2 1
2 1
1
C
C C
C C
C
Z
Z Z
Z Z
Z
C
Z (547)
] mol [m
0002551 , 0 0003580 , 0
0003580 , 0 0004853 , 0
2
2
1 - 3
2
3
3 / 1 3 / 1
3
3 / 1 3 / 1
1
2 1
2 1
(
=
(
(
(
(
(
(
|
|
.
|
\
|
+
|
|
.
|
\
|
+
=
C
C
v
v v
v v
v
C C
C C
C
v
(548)
(
=
(
(
(
(
(
=
0 01706 , 0
01706 , 0 0
0 1
1 0
12
2 1
12
2 1
C
C C
C
C C
v
v v
v
v v
C
k (549)
( )
( )
[K]
20 , 425 483,65
483,65 40 , 569
1
1
2 12 2 1
12 2 1 1
(
=
(
(
=
C C C
C C C
T k T T
k T T T
C
T (550)
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
168
[bar]
97 , 37 29,77
29,77 97 , 24
2
12
12 12
12
12 12
1
(
=
(
(
(
(
=
C
C
C C
C
C C
C
P
v
RT Z
v
RT Z
P
C
P (551)
(
=
(
(
(
(
+
+
=
1990 , 0 0,2985
0,2985 3980 , 0
2
2
2
1
j i
j i
(552)
(
=
41 , 1 24 , 1
24 , 1 05 , 1
R
T
(
=
42 , 0 54 , 0
54 , 0 64 , 0
R
P
(553)
(
=
(
=
0985 , 0 0694 , 0
0694 , 0 000947 , 0
1602 , 0 2159 , 0
2159 , 0 3051 , 0
1
o
B
B
(554)
(
=
(
=
41995 , 0 08456 , 0
08456 , 0 18530 , 0
14063 , 0 19517 , 0
19517 , 0 30472 , 0
RT
BP
RT
BP
C
C
(
=
(
=
12628 , 0 24491 , 0
24491 , 0 51733 , 0
29968 , 0 45564 , 0
45564 , 0 80799 , 0
R
P
dT
dB
dT
dB
(555)
= =
= =
N
i
N
j
ij
j i
RT
P B
x x
RT
BP
1 1
0991 , 0
= =
= |
.
|
\
|
=
N
i
N
j
ij
R j i R
P
dT
dB
x x P
dT
dB
1 1
28346 , 0
(556)
Si ottiene quindi:
9009 , 0 1 = +
RT
BP
Z (557)
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
169
In modo analogo si calcola la entalpia residua:
38256 , 0 28346 , 0 0991 , 0 = = |
.
|
\
|
=
dT
dB
T B
RT
P
RT
h
R
(558)
] mol [J 1908,40
1
= = RT
RT
h
h
R
R
(559)
I risultati ottenuti sono riassunti nella Tabella 13. Si nota che non vi sono grandi differenze
tra le diverse equazioni di stato in quanto il comportamento del fluido prossimo a quello
di un gas perfetto.
600 [K] e 16 [bar], gas 300 [K] e 3 [bar], liquido
EoS Z h
R
[J mol
-1
] Z h
R
[J mol
-1
]
Gas perfetto 1,00 0,0 1,000 0
VdW 0,89 -1416,5 0,028 -13000
RK 0,89 -1768,7 0,019 -25375
RKS 0,90 -2069,0 0,018 -31755
PR 0,89 -2165,2 0,016 -31092
Stati corrispondenti 0,91 -1991,7 0,016 -30657
Viriale 0,92 -1908,4 0,811 -2195
Tabella 13: valori dei coefficienti di compressibilit e delle entalpie residue calcolati con
le diverse EoS.
Seguendo lo stesso procedimento anche possibile calcolare il volume molare della
miscela equimolare a 300 [K] e 3 [bar]. In queste condizioni la miscela si trova in fase
liquida. Questa volta, come riportato in Tabella 13, le differenze sono marcate. In
particolare, le equazioni di stato del gas perfetto e del viriale forniscono ovviamente
risultati non corretti. Inoltre, anche lequazione di van der Waals fornisce risultati diversi da
quelli delle altre equazioni, che risultano viceversa abbastanza equivalenti.
Il valore dellentalpia molare della miscela si calcola poi sommando allentalpia residua il
valore dellentalpia molare della miscela nello stato di gas perfetto alla stessa temperatura.
Dalla relazione (456), prendendo come riferimento una temperatura T
r
=300 [K], si ottiene:
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
170
( )
( )
= =
+ +
+ + =
= = =
T
T
N
i
T
T
Pi i
N
i
i i
dT
dT
dT T C x T h x P T h
r
300
2 6 3
300
2 6 3
1
*
1
* *
T 10 97,796 - T 10 6,911 30 087 , 16 5 , 0
T 10 184,637 - T 10 586,694 900 , 67 5 , 0
) ( ) ( ) , , ( x
(560)
Risolvendo gli integrali si ottiene h*(300 [K]) = 0 e h*(600 [K]) = 64020 [J mol
-1
].
Come discusso nei capitoli precedenti, lo svolgimento di questo tipo di conti tedioso,
soprattutto se necessario ripeterli un gran numero di volte. Risulta quindi utile
implementare la sequenza dei calcoli in un programma di calcolo. A titolo di esempio, si
riporta di seguito un programma in linguaggio MATLAB, commentato e quindi di facile
lettura, che pu essere implementato in un file testo di nome vhsgTP_mix.m, con una
struttura simile al programma vhsgTP.m precedentemente discusso da cui derivato.
%vhsgTP calcolo del volume molare
% e delle funzioni residue
% a T e P con diverse EoS
% per miscele binarie
%
% R. Rota (2002)
format short g %formato scrittura
format compact %formato scrittura
clear all %cancella tutte le variabili
%dati per n-ottano
Tc(1) =569.4; %[K]
Pc(1) =24.97e5; %[Pa]
om(1) =0.398; %fattore acentrico di Pitzer
Zc(1) =0.256; %fattore di compressibilita' critico
%dati per n-butano
Tc(2) =425.2; %[K]
Pc(2) =37.97e5; %[Pa]
om(2) =0.199; %fattore acentrico di Pitzer
Zc(2) =0.274; %fattore di compressibilita' critico
%calcolo volume molare gas (fase=1) o liquido (fase=2)
fase = 1;
%assegno le condizioni di T e P e y1
T = 600; %temperatura [K]
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
171
P = 16e5; %pressione [Pa]
%T = 300; %temperatura [K]
%P = 3e5; %pressione [Pa]
y(1)= 0.5; %frazione molare del composto 1
y(2)= 1-y(1); %frazione molare del composto 2
R = 8.314; %[J/(mol K)]
RT =R*T;
RTc=R*Tc;
TR =T./Tc;
PR =P./Pc;
%1 gas perfetto
disp('gas perfetto')
v = R*T/P
hr=0
sr=0
gr=0
pause
%2 vdW, RK, RKS, PR
eq=['vdW';'RK ';'RKS';'PR ']; %nomi EoS
for tipo=1:4 %ciclo su diverse EoS
%calcolo Z a (P,T)
[Z,A,B,STrk] = EoS_mix(T,P,y,Tc,Pc,om,tipo);
disp(eq(tipo,:)) %visualizzo nome EoS
if fase == 1
Z(3) %visualizzo Z gas
v = Z(3)*RT/P %calcolo Vgas [m3/mol]
[hrRT,srR] = fr(Z(3),A,B,STrk,tipo) %calc. hr/RT e sr/R
hr = hrRT*RT
sr = srR*R
gr = hr - T*sr
else
Z(1) %visualizzo Z liquido
v = Z(1)*RT/P %calcolo Vliq [m3/mol]
[hrRT,srR] = fr(Z(1),A,B,STrk,tipo); %calc. hr/RT e sr/R
hr = hrRT*RT
sr = srR*R
gr = hr - T*sr
end
pause
end
%3 viriale
disp('viriale') %visualizzo nome EoS
vc =Zc.*R.*Tc./Pc;
for i=1:2
for j=1:2
vcm(i,j)=((vc(i)^(1/3)+vc(j)^(1/3))/2)^3;
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
172
Zcm(i,j)= mean([Zc(i), Zc(j)]);
kc(i,j) =1-sqrt(vc(i)*vc(j))/vcm(i,j);
Tcm(i,j)=sqrt(Tc(i)*Tc(j))*(1-kc(i,j));
omm(i,j)= mean([om(i), om(j)]);
end
end
Pcm=Zcm.*R.*Tcm./vcm;
TRm=T./Tcm;
PRm=P./Pcm;
B0ij = 0.083-0.422./TRm.^1.6 ; %matrice B0ij
B1ij = 0.139-0.172./TRm.^4.2 ; %matrice B1ij
BijPRT = (B0ij+omm.*B1ij).*PRm./TRm; %matrice Bij*(P/RT)
BPRT = y*BijPRT*y'; %B*(P/RT) di miscela
Z = 1+BPRT %calcolo Z
dBdTijPR = (0.675./TRm.^2.6+omm*0.722./TRm.^5.2).*PRm; %matrice
dB/dTij*(P/R)
dBdTPR = y*dBdTijPR*y'; %calcolo dB/dT*(P/R) di miscela
hrRT =BPRT - dBdTPR;
srR =- dBdTPR;
v = Z*RT/P %calcolo v [m3/mol]
hr = hrRT*RT
sr = srR*R
gr = hr - T*sr
Il programma vhsgTP_mix.m chiama due sottoprogrammi, EoS_mix.m e fr.m. Il primo
ha funzioni analoghe al sottoprogramma EoS.m (da cui derivato con poche modifiche)
discusso per il caso di composti puri. Il secondo gi stato discusso per il caso di composti
puri.
function [Z,A,B,STrk] = EoS_mix(T,P,y,Tc,Pc,om,tipo)
%EoS_mix calcola Z con diverse EoS per una miscela binaria
% senza coeff. di interazione binaria
% dati T [K], P [Pa], y [-], Tc [K],
% Pc [Pa], om [-], tipo (EoS da usare)
% tipo: 1 vdW
% 2 RK
% 3 RKS
% 4 PR
% restituisce Z (ordinato) oppure Z, A, B, S e k
%
% uso: Z = EoS(T,P,y,Tc,Pc,om,tipo)
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
173
% [Z,A,B,STrk] = EoS(T,P,y,Tc,Pc,om,tipo)
%
% R. Rota (2002)
R =8.314; %costante gas perfetti [J/(mol K)]
RT =R*T;
RTc=R*Tc;
TR =T./Tc;
%parametri dell'equazione nalla forma Z^3+alfa*Z^2+beta*Z+gamma=0
if tipo == 1 %vdW
S =[0 0]; %calcolo vettore riga dei coeff. S dei puri
k =[1 1]; %calcolo vettore riga dei coeff. k dei puri
%calcolo vettore riga dei coeff. a dei puri
a =27*RTc.^2 ./ (64*Pc);
%calcolo vettore riga dei coeff. b dei puri
b =RTc ./ (8*Pc);
aM =sqrt(a'*a); %calcolo matrice dei coeff. aij
am =y*aM*y'; %calcolo a di miscela
bm =b*y'; %calcolo b di miscela
A =am*P/(RT)^2;
B =bm*P/(RT);
alfa =-1-B;
beta =A;
gamma=-A*B;
end
if tipo ==2 %RK
S =[0 0]; %calcolo vettore riga dei coeff. S dei puri
k =[1 1]; %calcolo vettore riga dei coeff. k dei puri
TR =T./Tc;
%calcolo vettore riga dei coeff. a dei puri
a =0.42748*RTc.^2 ./ (Pc.*sqrt(TR));
%calcolo vettore riga dei coeff. b dei puri
b =0.08664*RTc ./ Pc;
aM =sqrt(a'*a); %calcolo matrice dei coeff. aij
am =y*aM*y'; %calcolo a di miscela
bm =b*y'; %calcolo b di miscela
A =am*P/(RT)^2;
B =bm*P/(RT);
alfa =-1;
beta =A-B-B^2;
gamma=-A*B;
end
if tipo ==3 %RKS
%calcolo vettore riga dei coeff. S dei puri
S =0.48+1.574*om-0.176*om.^2;
%calcolo vettore riga dei coeff. k dei puri
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
174
k =(1+S.*(1-sqrt(T./Tc))).^2;
%calcolo vettore riga dei coeff. a dei puri
a =0.42748 * k .* RTc.^2 ./ Pc;
%calcolo vettore riga dei coeff. b dei puri
b =0.08664 * RTc ./ Pc;
aM =sqrt(a'*a); %calcolo matrice dei coeff. aij
am =y*aM*y'; %calcolo a di miscela
bm =b*y'; %calcolo b di miscela
A =am*P/(RT)^2;
B =bm*P/(RT);
alfa =-1;
beta =A-B-B^2;
gamma=-A*B;
end
if tipo == 4 %PR
%calcolo vettore riga dei coeff. S dei puri
S =0.37464+1.54226*om-0.26992*om.^2;
%calcolo vettore riga dei coeff. k dei puri
k =(1+S.*(1-(T./Tc).^0.5)).^2;
%calcolo vettore riga dei coeff. a dei puri
a =0.45724 * k .* RTc.^2 ./ Pc;
%calcolo vettore riga dei coeff. b dei puri
b =0.07780 * RTc ./ Pc;
aM =sqrt(a'*a); %calcolo matrice dei coeff. aij
am =y*aM*y'; %calcolo a di miscela
bm =b*y'; %calcolo b di miscela
A =am*P/(RT)^2;
B =bm*P/(RT);
alfa =-1+B;
beta =A-2*B-3*B^2;
gamma=-A*B+B^2+B^3;
end
%calcolo parametro per funzioni residue
STrk=sum(y.*S.*sqrt(a.*TR./k))./sqrt(am);
%risoluzione analitica della cubica
p = beta-alfa^2/3;
q = 2*alfa^3/27-alfa*beta/3+gamma;
q2=q/2;
a3=alfa/3;
D = q^2/4+p^3/27;
if D > 0
Z1 = (-q2+sqrt(D))^(1/3)+(-q2-sqrt(D))^(1/3)-a3;
Z = [Z1 Z1 Z1]
elseif D == 0
Z1 = -2*(q2)^(1/3)-a3;
Z2 = (q2)^(1/3)-a3;
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
175
Z = [Z1 Z2 Z2];
else
r = sqrt(-p^3/27);
teta= acos(-q2*sqrt(-27/p^3));
Z1 = 2*r^(1/3)*cos(teta/3)-a3;
Z2 = 2*r^(1/3)*cos((2*pi+teta)/3)-a3;
Z3 = 2*r^(1/3)*cos((4*pi+teta)/3)-a3;
Z = [Z1 Z2 Z3];
end
Z =sort(Z); %ordina i valori
5.5 Potenziale chimico e fugacit
Analogamente al caso dei fluidi puri, anche per le miscele si presenta la necessit di poter
correlare il potenziale chimico di un composto nella miscela a delle variabili misurabili per
definire le condizioni di equilibrio al trasferimento di materia. Sempre seguendo
lapproccio di Lewis, si introduce la fugacit anche per un composto in una miscela (nel
seguito indicata col simbolo
i
f
per distinguerla dalla fugacit del composto puro), definita
in termini differenziali per analogia alla relazione (468) ricavata per una miscela di gas
perfetti:
i T i
f RTd d
ln
,
= (561)
Poich risulta evidente che la fugacit di un composto in una miscela di gas perfetti
coincide con la pressione parziale, ricordando che tutti i composti per pressioni
sufficientemente basse si comportano come gas perfetti, la definizione precedente viene
completata dalla seguente:
1
lim
0 P
=
|
|
.
|
\
|
i
i
P
f
(562)
Per un composto in miscela, il rapporto tra la fugacit e la pressione parziale viene
solitamente chiamato coefficiente di fugacit,
i
, e la relazione precedente diviene quindi:
( ) 1
lim
0
=
i
P
(563)
Come per il caso dei fluidi puri, il coefficiente di fugacit rappresenta il rapporto tra la
fugacit di un composto in una miscela e la fugacit del composto in una miscela di gas
perfetti nelle stesse condizioni:
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
176
( )
( )
( )
( )
i
i
i
i
i
Px
P T f
P T f
P T f
P T
x
x
x
x
, ,
, ,
, ,
, ,
*
= = (564)
Risulta evidente che il coefficiente di fugacit di un composto in una miscela di gas perfetti
uguale a uno.
Per un fluido reale viceversa
i
risulta diverso da uno e pu essere correlato allenergia
libera residua (in questo caso parziale molare) e calcolato, analogamente al caso dei fluidi
puri, utilizzando una EoS per miscele. Infatti, integrando la relazione (561) tra una
condizione di gas perfetto e una di fluido reale, si ottiene:
|
|
.
|
\
|
= =
i
i
i i
P T
P T
i
P T
P T
i
P
f
RT P T P T f d RT d
ln ) , , ( ) , , (
ln
*
, ,
,* , ,
, ,
,* , ,
x x
x
x
x
x
(565)
Ricordando che il potenziale chimico di un composto in miscela coincide con lenergia
libera di Gibbs parziale molare e utilizzando la definizione di funzione residua, la relazione
precedente pu essere riscritta come:
) , , (
ln ) , , ( ) , , ( ) , , (
*
x x x x P T RT P T G P T G P T G
i
R
i i i
= = (566)
Ne consegue che, analogamente al caso dei fluidi puri, il coefficiente di fugacit di un
composto in una miscela pu essere calcolato dallenergia libera di Gibbs residua, anche se
per il caso di una miscela richiesta unoperazione di derivazione. Infatti, utilizzando la
definizione di grandezza parziale molare, si ottiene:
( )
i j
n P T
i
R
i
n
RT P T G
P T
|
|
.
|
\
|
=
, ,
) , , (
) , , (
ln
x
x
(567)
Dalla relazione (273) si ottiene:
( ) ( )
( ) cost , 1
, , , ,
0
= = =
n T
P
dP
Z n
RT
P T ng
RT
P T G
P
R R
x x
(568)
e quindi risulta:
( )
i j
n P T
i
P
i
n
P
dP
Z n
P T
|
|
|
|
|
.
|
\
|
|
.
|
\
|
=
, ,
0
1
) , , (
ln x
(569)
da cui si ottiene:
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
177
( )
|
|
|
.
|
\
|
|
|
.
|
\
|
=
|
|
.
|
\
|
=
P
n P T
i
P
n P T
i
i
P
dP
n
nZ
P
dP
n
Z n
P T
i j i j
0 , , 0 , ,
1
1
) , , (
ln x (570)
Come nel caso dei fluidi puri, se nota una EoS possibile effettuare la derivazione e poi
lintegrazione contenuta nellequazione precedente e quindi calcolare il valore del
coefficiente di fugacit. Si noti che la dipendenza di Z da n
i
contenuta nelle regole di
miscelazione discusse in precedenza.
Una notevole differenza rispetto al caso dei fluidi puri risiede nel fatto che nel caso di
miscele Z non dipende solo dalle variabili ridotte ma anche dalla composizione. Questo
implica che non possibile dedurre delle relazioni generalizzate per il calcolo del
coefficiente di fugacit valide per tutte le miscele in funzione delle sole variabili ridotte.
Si possono invece utilizzare diverse EoS per calcolare il coefficiente di fugacit con la
relazione precedente
96
.
In particolare, utilizzando lEoS del viriale troncata al secondo coefficiente:
RT
BP
Z + =1 (571)
con le regole di miscelazione (478):
= =
=
N
i
N
j
ij j i
B x x B
1 1
(572)
facile calcolare analiticamente la derivata e lintegrale della (569):
( ) ( )
( )
( )
( )
=
= =
+ + =
=
|
|
|
.
|
\
|
+ =
|
|
.
|
\
|
+ =
=
|
|
.
|
\
|
+
|
|
.
|
\
|
=
|
|
.
|
\
|
+
=
|
|
.
|
\
|
N
j
ij j
n P T
i
N
i
N
j
ij j i
n P T
i
n P T
i
n P T
i
n P T
i
n P T
i
B x B
RT
P
n
n B n n
RT
P
n
nB
RT
P
n
RT BP n
n
n
n
RT BP n
n
nZ
i j
i j
i j i j i j i j
1
, ,
1 1
, ,
, , , , , , , ,
2 1
1 1
1
(573)
da cui:
96
Come nel caso dei fluidi puri, lequazione precedente non utilizzabile se lEoS non fornisce Z
come una funzione esplicita di P. Quindi lequazione precedente utilizzabile con lEoS del viriale,
mentre per le EoS cubiche deve essere rimaneggiata come discusso per esempio nel testo H.C. van
Ness e M.M. Abbott Classical thermodynamics of nonelectrolyte solutions, McGraw Hill, 1982.
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
178
|
.
|
\
|
+ =
= |
.
|
\
|
|
.
|
\
|
+ + =
=
|
|
|
.
|
\
|
|
|
.
|
\
|
=
=
N
j
ij j
P
N
j
ij j
P
n P T
i
i
B x B
RT
P
P
dP
B x B
RT
P
P
dP
n
nZ
P T
i j
1
0
1
0 , ,
2
1 2 1
1 ) , , (
ln x
(574)
Come discusso per il caso dei fluidi puri, possibile ricavare espressioni analoghe anche
utilizzando le EoS cubiche. In questo caso il coefficiente di fugacit risulta dalle relazioni
riassunte in Tabella 14.
Poich le equazioni di stato cubiche sono in grado di rappresentare il comportamento sia
della fase vapore sia di quella liquida, possibile utilizzarle per calcolare il coefficiente di
fugacit di entrambe le fasi. La procedura di calcolo in questo caso la seguente:
assegnare temperatura, pressione e composizione della miscela e calcolare i parametri
A
i
e B
i
dei composti puri e, tramite le regole di miscelazione, i parametri A e B delle
miscele;
risolvere la cubica in Z e selezionare la radice minore (se si vuole calcolare il
coefficiente di fugacit del liquido) o quella maggiore (se si vuole calcolare il
coefficiente di fugacit del vapore);
calcolare il coefficiente di fugacit della specie i nella miscela con la relazione
opportuna.
EoS
i
ln
RK ( ) ( ) B Z
Z
B Z
A
A
B
B
B
A
Z
B
B
i i i
|
.
|
\
| +
|
|
.
|
\
|
+ ln ln 2 1
RKS ( ) ( ) B Z
Z
B Z
A
A
B
B
B
A
Z
B
B
i i i
|
.
|
\
| +
|
|
.
|
\
|
+ ln ln 2 1
PR ( )
( )
( )
( ) B Z
B Z
B Z
A
A
B
B
B
A
Z
B
B
i i i
|
|
.
|
\
|
+
+ +
|
|
.
|
\
|
+ ln
2 1
2 1
ln 2
2 2
1
Tabella 14: espressioni per il calcolo del coefficiente di fugacit di composti puri con le
EoS cubiche pi utilizzate.
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
179
5.5.1 Applicazione
Si calcoli il coefficiente di fugacit dei due componenti una miscela equimolare di n-ottano
e n-butano a 600 [K] e 16 [bar] in fase vapore con lEoS RK trascurando i parametri di
interazione binaria nelle regole di miscelazione.
Procedendo esattamente come illustrato per il caso dei composti puri si ricavano per prima
cosa i parametri relativi ai composti puri, che assumono i seguenti valori:
n-butano (1) n-ottano (2)
a [m
6
Pa mol
-2
] 1,1844 3,7375
b [m
3
mol
-1
] 8,0664 10
-5
1,6426 10
-4
A 0,076155 0,24032
B 0,025873 0,052685
Utilizzando questi valori si ricavano i parametri per la miscela:
] mol Pa [m 2855 , 2
2 6
2
1
2
1
= =
= =
i j
j i j i
a a x x a (575)
] mol [m 10 2246 , 1
1 3 4
2
1
=
= =
i
i i
b x b (576)
( )
0,14676
2
= =
RT
aP
A
(577)
0,039279 = =
RT
bP
B (578)
da cui, risolvendo lequazione cubica in Z esattamente come per il caso dei fluidi puri, si
ricava il valore del coefficiente di compressibilit della miscela vapore:
88801 , 0 = Z (579)
Il valore dei coefficienti di attivit dei due composti si ricavano con la relazione:
( ) ( )
=
|
|
.
|
\
|
|
.
|
\
| +
|
|
.
|
\
|
+ =
0,83263
0,96443
ln ln 2 1 exp
B Z
Z
B Z
A
A
B
B
B
A
Z
B
B
i i i
i
(580)
da cui si vede come il comportamento del n-butano sia ben approssimabile come quello di
un gas perfetto, mentre non lo quello del n-ottano.
Come dovrebbe essere ormai chiaro, risulta utile implementare la sequenza dei calcoli in un
programma di calcolo. A titolo di esempio, si riporta di seguito un programma in
linguaggio MATLAB, come al solito commentato e quindi di facile lettura, che pu essere
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
180
implementato in un file testo di nome phi_mix.m, con una struttura simile al programma
vhsgTP_mix.m precedentemente discusso da cui derivato.
%phi calcolo del coefficiente di
% fugacit a T, P e y con diverse EoS
% per miscele binarie
%
% R. Rota (2002)
format short g %formato scrittura
format compact %formato scrittura
clear all %cancella tutte le variabili
%dati per n-ottano
Tc(1) =569.4; %[K]
Pc(1) =24.97e5; %[Pa]
om(1) =0.398; %fattore acentrico di Pitzer
Zc(1) =0.256; %fattore di compressibilita' critico
%dati per n-butano
Tc(2) =425.2; %[K]
Pc(2) =37.97e5; %[Pa]
om(2) =0.199; %fattore acentrico di Pitzer
Zc(2) =0.274; %fattore di compressibilita' critico
%calcolo volume molare gas (fase=1) o liquido (fase=2)
fase = 1;
%assegno le condizioni di T e P e y1
T = 600; %temperatura [K]
P = 16e5; %pressione [Pa]
y(1)= 0.5; %frazione molare del composto 1
y(2)= 1-y(1); %frazione molare del composto 2
R = 8.314; %cost. gas [J/(mol K)]
RT =R*T;
RTc=R*Tc;
TR =T./Tc;
PR =P./Pc;
%1 gas perfetto
disp('gas perfetto')
phi = [1 1 ]
pause
%2 RK, RKS, PR
eq=['vdW';'RK ';'RKS';'PR ']; %nomi EoS
for tipo=2:4 %ciclo su diverse EoS
%calcolo A e B per i composti puri
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
181
for j=1:2,
[Z,Ap(j),Bp(j),S,k] = EoS(T,P,Tc(j),Pc(j),om(j),tipo);
end
%calcolo Z a (P,T,y)
[Z,A,B,STrk] = EoS_mix(T,P,y,Tc,Pc,om,tipo);
disp(eq(tipo,:)) %visualizzo nome EoS
if fase == 1
Zm=Z(3); %Z vapore
else
Zm=Z(1); %Z liquido
end
logphi = lnphi(Ap,Bp,A,B,Zm,tipo);
phi = exp(logphi)
pause
end
%3 viriale
disp('viriale') %visualizzo nome EoS
vc =Zc.*R.*Tc./Pc;
for i=1:2
for j=1:2
vcm(i,j)=((vc(i)^(1/3)+vc(j)^(1/3))/2)^3;
Zcm(i,j)= mean([Zc(i), Zc(j)]);
kc(i,j) =1-sqrt(vc(i)*vc(j))/vcm(i,j);
Tcm(i,j)=sqrt(Tc(i)*Tc(j))*(1-kc(i,j));
omm(i,j)= mean([om(i), om(j)]);
end
end
Pcm=Zcm.*R.*Tcm./vcm;
TRm=T./Tcm;
PRm=P./Pcm;
B0ij = 0.083-0.422./TRm.^1.6 ; %matrice B0ij
B1ij = 0.139-0.172./TRm.^4.2 ; %matrice B1ij
BijPRT = (B0ij+omm.*B1ij).*PRm./TRm; %matrice Bij*(P/RT)
BPRT = y*BijPRT*y'; %B*(P/RT) di miscela
logphi=-BPRT+2*y*BijPRT;
phi = exp(logphi)
Il programma phi_mix.m chiama due sottoprogrammi, EoS_mix.m e EoS.m gi discussi
in precedenza.
Con questo programma facile ripetere i calcoli utilizzando diverse EoS o diverse
condizioni, come riportato in Tabella 15. Seguendo lo stesso procedimento anche
possibile calcolare i coefficienti di fugacit della miscela equimolare a 300 [K] e 3 [bar]. In
queste condizioni la miscela si trova in fase liquida.
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
182
600 [K] e 16 [bar], gas 300 [K] e 3 [bar], liquido
EoS
1
ln
2
ln
1
ln
2
ln
Gas perfetto 1,00 1,00 1,000 1,000
RKS 0,98 0,84 0,810 0,007
PR 0,97 0,82 0,806 0,007
Viriale 0,97 0,84 0,958 0,655
Tabella 15: valori dei coefficienti di fugacit calcolati con le diverse EoS.
5.6 Fasi condensate
Come discusso per il caso dei fluidi puri, luso di equazioni di stato in grado di
rappresentare il comportamento volumetrico anche della fase liquida consente il calcolo
delle funzioni residue, e quindi del valore di tutte le propriet, anche per la fase liquida. Si
anche detto in precedenza che le equazioni di stato, sviluppate come correzioni sempre pi
accurate dellEoS del gas perfetto, sono generalmente in grado di rappresentare meglio il
comportamento della fase gas rispetto a quello della fase liquida. Di conseguenza, anche il
valore delle funzioni residue calcolate con le relazioni discusse nella sezione precedente
sono pi affidabili per la fase gas che non per la fase liquida.
Un approccio alternativo (chiamato, come per il caso dei composti puri, metodo indiretto
per contrasto con quello che utilizza direttamente una EoS e viene per questo chiamato
metodo diretto) per il calcolo delle funzioni termodinamiche che non richiede lutilizzo
delle equazioni di stato per la fase condensata quello che sfrutta delle ulteriore
informazione sperimentali come descritto nel seguito.
5.6.1 Miscele ideali
La pi semplice soluzione liquida che si possa immaginare quella per cui, analogamente a
quanto avviene per una miscela di gas perfetti, le interazioni steriche ed energetiche tra le
molecole dei composti puri sono identiche a quelle tra le diverse molecole nella miscela.
Nel caso di gas perfetti tali interazioni sono nulle sia per i composti puri sia per le miscele,
mentre nel caso di liquidi tali interazioni non possono essere nulle in quanto devono
esistere delle forze attrattive responsabili della condensazione. Se per le interazioni tra
molecole uguali e molecole diverse sono le stesse, il volume e lentalpia di miscelazione
(come per il caso di miscele di gas perfetti) dovranno essere nulli. Come discusso per il
caso di una miscela di gas perfetti, questo significa che il processo di miscelazione non
associato a una variazione del volume ed atermico. Questa approssimazione ovviamente
tanto pi ragionevole quanto pi i composti sono simili e quanto pi le interazioni tra le
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
183
molecole sono deboli
97
. Sempre analogamente a quanto avviene per il gas perfetto,
lentropia di miscelazione non sar viceversa nulla ma positiva, cio il processo di
miscelazione sar sempre comunque spontaneo.
Tutto questo risulta verificato se lenergia libera di Gibbs parziale molare dipende dalla
composizione in modo analogo a quella di una miscela di gas perfetti, equazione (465):
i i i
x RT P T g P T G ln ) , ( ) , , ( + =
x (581)
Le miscele per cui vale la relazione precedente sono chiamate miscele ideali, le cui
propriet saranno indicate con lapice
. La relazione precedente differisce dalla (465) in
quanto lenergia libera molare del composto puro non quella nello stato di gas perfetto ma
nel reale stato in cui il composto puro si trova a T e P, per esempio in fase liquida
98
. In
questo modo tutta la differenza tra il gas perfetto e la fase condensata (che nelle funzioni
residue viene valutata attraverso una EoS) viene compresa nel valore dellenergia libera di
Gibbs dei composti puri, che per le fasi condensate pu essere calcolata dai valori
dellentalpia e dellentropia fruendo dellinformazione sperimentale relativa allentalpia di
evaporazione (come discusso nella sezione 4.5). Questo il principale vantaggio di questo
approccio rispetto alluso diretto di unEoS.
Le altre grandezze parziali molari si ricavano immediatamente dalla (581) attraverso le
relazioni (442) e (441):
i i i
P
i
P
i
i
x R P T s x R
T
g
T
G
P T S ln ) , ( ln ) , , (
,
,
=
|
|
.
|
\
|
=
|
|
.
|
\
|
x
x
x
(582)
) , ( ) , , (
,
,
P T v
P
g
P
G
P T V
i
T
i
T
i
i
=
|
|
.
|
\
|
=
|
|
.
|
\
|
x
x
x
(583)
( )
) , ( ) , ( ) , (
ln ) , ( ln ) , (
) , , ( ) , , ( ) , , (
P T h P T Ts P T g
x R P T s T x RT P T g
P T S T P T G P T H
i i i
i i i i
i i i
= + =
= + + =
= + =
x x x
(584)
97
Lapprossimazione di miscela ideale sar quindi ragionevole per una miscela m-xilene / p-xilene,
ma non per una miscela acqua / toluene.
98
La trattazione delle miscele ideali stata inserita nella sezione fasi condensate in quanto
rappresenta il punto di partenza per la trattazione pi comune delle miscele liquide. Bisogna per
sottolineare che molte miscele gassose si comportano come miscele ideali, nonostante il
comportamento della miscela e dei singoli gas puri non sia quello di un gas perfetto. In questo caso, la
propriet del fluido puro sar ovviamente quella di un gas reale, caratterizzabile con una EoS per i
fluidi puri. Risulta inoltre evidente che tutte le miscele di gas perfetti sono anche miscele ideali.
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
184
) , ( ) , ( ) , (
) , , ( ) , , ( ) , , (
P T u P T Pv P T h
P T V P P T H P T U
i i i
i i i
= =
= =
x x x
(585)
Queste relazioni risultano identiche a quelle ricavate per una miscela di gas perfetti con la
sola differenza che le propriet dei fluidi puri sono relative non a un gas perfetto ma al
fluido reale a T e P.
Ne consegue che anche le propriet della miscela si calcolano con relazioni molto semplici,
analoghe a quelle dedotte per il caso di una miscela di gas perfetti:
= = =
= = =
=
= =
= =
+ = =
= =
=
=
= =
= =
N
i
i i
N
i
i i
N
i
i i
N
i
i i
N
i
i i
N
i
i i
N
i
i i V V
N
i
i i P P
N
i
i i
N
i
i i
N
i
i i
N
i
i i
x x RT x P T g x P T G P T g
x x R x P T s x P T S P T s
x P T c P T c
x P T c P T c
x P T u x P T U P T u
x P T h x P T H P T h
1 1 1
1 1 1
1
,
1
,
1 1
1 1
ln ) , ( ) , , ( ) , , (
ln ) , ( ) , , ( ) , , (
) , ( ) , , (
) , ( ) , , (
) , ( ) , , ( ) , , (
) , ( ) , , ( ) , , (
x x
x x
x
x
x x
x x
(586)
Anche queste relazioni risultano identiche a quelle ricavate per una miscela di gas perfetti
con la sola differenza che le propriet dei fluidi puri sono relative non a un gas perfetto ma
al fluido reale nelle condizioni di T e P, siano esse quelle di un gas reale o di un liquido.
Ancora una volta, il principale vantaggio di questo approccio consiste nel poter fruire, per il
calcolo delle propriet di una fase condensata, dellinformazione sperimentale relativa
allentalpia di evaporazione (come discusso nella sezione 4.5).
Il calcolo delle propriet di una miscela ideale risulta quindi, come per il caso delle miscele
di gas perfetti, estremamente semplice in quanto sufficiente conoscere le propriet dei
fluidi puri nelle stesse condizioni di temperatura e pressione della miscela (liquida o
gassosa che sia) oltre alla composizione della miscela.
5.6.1.1 Propriet di miscelazione
Come discusso per il caso di miscele di gas perfetti, quando dei fluidi puri vengono
miscelati, le propriet della miscela che si forma possono essere diverse dalla somma delle
propriet dei fluidi puri di partenza di una quantit pari, per definizione, alla propriet di
miscelazione.
Dalle relazioni ricavate in precedenza per le diverse propriet di una miscela ideale si pu
ricavare che, esattamente come per il caso di una miscela di gas perfetti, il volume,
lentalpia e lenergia interna molare di miscelazione sono nulli, come logico attendersi
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
185
poich le interazioni steriche ed energetiche tra le molecole dello stesso composto e di
composti diversi sono uguali:
0 ) , ( ) , , ( ) , , (
1
,
= =
=
N
i
i i
mix
P T h x P T h P T h x x (587)
0 ) , ( ) , , ( ) , , (
1
,
= =
=
N
i
i i
mix
P T u x P T u P T u x x (588)
0 ) , ( ) , , ( ) , , (
1
,
= =
=
N
i
i i
mix
P T v x P T v P T v x x (589)
Come per il caso di miscele di gas perfetti, il fatto che il volume di miscelazione sia nullo
significa che miscelando 1 litro del composto A con 1 litro del composto B si ottengono 2
litri di miscela. Il processo di miscelazione non cio associato a una variazione del
volume.
Il fatto che lenergia interna (o lentalpia) di miscelazione sia nulla significa che miscelando
senza scambiare energia con lambiente a volume (o pressione) costante il composto A a 20
[C] con il composto B a 20 [C] si ottiene una miscela a 20 [C]. Il processo di
miscelazione cio atermico.
Viceversa, anche per le miscele ideali lentropia molare di miscelazione diversa da zero e
pari a:
0 ln
) , ( ln ) , (
) , ( ) , , ( ) , , (
1
1
1 1
1
,
> =
= =
= =
=
=
= =
=
N
i
i i
N
i
i i
N
i
i i
N
i
i i
N
i
i i
mix
x x R
P T s x x x R P T s x
P T s x P T s P T s x x
(590)
Questo mostra come, analogamente al caso di miscele di gas perfetti, alla miscelazione
associato un aumento di entropia ed quindi un processo spontaneo.
5.6.1.2 Fugacit
Anche il calcolo della fugacit dei composti in miscele ideali risulta particolarmente
semplice. Infatti, integrando la relazione (395) tra lo stato di composto puro a T e P e lo
stato di composto in una miscela ideale a T, P e x si ottiene:
=
, , ,
,
, , ,
,
ln
x x P T
P T
i
P T
P T
i
f RTd d
(591)
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
186
( ) ( )
( )
( )
|
|
.
|
\
|
=
P T f
P T f
RT P T P T
i
i
i i
,
, ,
ln , , ,
x
x
(592)
Questultima relazione, ricordando che il potenziale chimico coincide con lenergia libera
di Gibbs parziale molare che, a sua volta, per un composto puro coincide con lenergia
libera di Gibbs molare, pu essere riscritta nella forma:
( ) ( )
( )
( )
|
|
.
|
\
|
=
P T f
P T f
RT P T g P T G
i
i
i i
,
, ,
ln , , ,
x
x
(593)
Confrontando questa relazione con la definizione di miscela ideale (581) si ricava:
( ) ( )
i i i
x P T f P T f , , ,
x (594)
questa relazione, detta di Lewis - Randall
99
, consente il calcolo della fugacit del composto
in miscela sulla base del valore della fugacit del composto puro nella stessa fase e nelle
stesse condizioni di T e P della miscela (valutabile come discusso nei capitoli precedenti) e
della composizione.
Dalla definizione di coefficiente di fugacit di un composto in miscela si ricava poi che:
( )
( ) ( ) ( )
( ) P T
P
P T f
Px
x P T f
P
P T f
P T
i
i
i
i i
i
i
i
,
, , , ,
, ,
= = = =
x
x (595)
Cio, il coefficiente di fugacit del composto in miscela coincide con quello del composto
puro alla temperatura e pressione della miscela e pu quindi essere calcolato come discusso
nel capitolo precedente.
5.6.1.3 Applicazione
Si calcoli la differenza di entalpia tra una miscela al 70% di n-ottano e 30% di n-butano in
fase vapore a 450 [K] e 1 [bar] e la stessa miscela in fase liquida a 300 [K] e 10 [bar]
considerando ideali entrambe le miscele.
I dati relativi al n-ottano puro sono gi stati utilizzati nellApplicazione 4.5.1 e sono
riassunti nella tabella seguente insieme ai dati relativi al n-butano. Nella stessa tabella sono
anche riportati i valori delle entalpie molari residue per i composti in fase vapore in diverse
condizioni, calcolati utilizzando lEoS PR come discusso nel capitolo precedente.
99
Merle Randall, 1888 1950, chimico statunitense.
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
187
n-butano (1) n-ottano (2)
temperatura di ebollizione normale, T
ev
[K] 272,7 398,8
entalpia di evaporazione normale, h
ev
[kJ mol
-1
] 22,500 34,799
calore specifico del liquido,
L
P
c [J mol
-1
K
-1
]
480 330
h
R,Vs
(T
ev
) [J mol
-1
] -210 -452
h
R,V
(450 [K], 1 [bar]) [J mol
-1
] -98 -365
La differenza di entalpia tra i due stati dei composti puri si calcola come discusso
nellApplicazione 4.5.1, da cui la differenza di entalpia della miscela tra i due stati A (fase
vapore a 450 [K] e 1 [bar]) e B (fase liquida a 300 [K] e 10 [bar]) si calcola come:
( )
( ) ( ) ( )
( ) ( )
( ) ( ) ( )
=
=
=
= =
(
(
+ + =
=
(
(
(
|
|
|
.
|
\
|
+ +
+
|
|
|
.
|
\
|
+ + + =
= =
= =
= =
2
1
,
, , ,
, *
, * *
2
1
, , ,
, * *
2
1
2
1
2
1
,
,
) , ( ) , (
) , ( ) , (
) , , ( ) , , (
,
,
,
,
i
A A
V R
i
T
T
L
Pi i ev i ev i ev
Vs R
i
T
T
Pi i
A A
V R
i
T
T
Pi r i
i
T
T
L
Pi i ev i ev i ev
Vs R
i
T
T
Pi r i i
i
A A
V
i B B
L
i i
i
A A
V
i i
i
B B
L
i i
A A
V
B B
L
B A
P T h dT c T h T h dT c x
P T h dT c T h
dT c T h T h dT c T h x
P T h P T h x
P T h x P T h x
P T h P T h h
B
i ev
i ev
A
A
r
B
i ev
i ev
r
x x
(596)
( ) ( )
( )
( ) ( )
( ) ] mol [J 66321 365 330 34799
452 10 637 , 184 10 694 , 586 900 , 67 7 , 0
98 480 22500
210 10 796 , 97 10 911 , 306 087 , 16 3 , 0
1
300
8 , 398
8 , 398
450
2 6 3
300
7 , 272
7 , 272
450
2 6 3
= +
+ + +
+ +
+ + + =
dT
dT T T
dT
dT T T h
B A
(597)
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
188
5.6.2 Miscele non ideali
La trattazione delle miscele non ideali, cio di quelle miscele che non seguono lequazione
(581), si sviluppa esattamente analoga a quella dei fluidi reali, cio di quei fluidi che non
seguono lequazione di stato dei gas perfetti.
Cos come nel caso dei fluidi reali si definiscono le funzioni residue come differenza tra il
valore della propriet di un fluido reale e quella di un gas perfetto nelle stesse condizioni,
nel caso di miscele reali si definiscono le funzioni di eccesso, solitamente indicate con
lapice
E
, come la differenza tra il valore della propriet di una miscela e quella di una
miscela ideale nelle stesse condizioni
100
. Per una generica grandezza m, si definisce quindi
funzione di eccesso:
( ) ( ) ( ) x x x , , , , , , P T m P T m P T m
E
= (598)
Risulta evidente che, poich i valori delle funzioni termodinamiche di una miscela ideale
sono facilmente calcolabili come discusso nelle sezioni precedenti, dal valore della
funzione di eccesso si pu facilmente risalire al valore della grandezza.
Si in precedenza definito il coefficiente di fugacit di un composto in una miscela come il
rapporto tra la fugacit del composto in una miscela e la fugacit del composto in una
miscela di gas perfetti nelle stesse condizioni. Analogamente, per le fasi condensate risulta
utile definire il coefficiente di attivit di un composto in una miscela, solitamente indicato
con
i
, come il rapporto tra la fugacit del composto in una miscela e la fugacit del
composto in una miscela ideale nelle stesse condizioni:
( )
( )
( )
( )
( )
i
L
i
L
i
L
i
L
i
i
x P T f
P T f
P T f
P T f
P T
,
, ,
, ,
, ,
, ,
x
x
x
x = =
(599)
in cui si inserita anche la relazione di Lewis Randall. Evidentemente, il coefficiente di
attivit in una miscela ideale uguale a uno. La relazione precedente mostra per anche che
quando la frazione molare di un composto tende a uno (cio la miscela tende verso un
composto puro) il relativo coefficiente di attivit diviene unitario:
( )
( )
( )
( )
( )
1
,
,
,
, ,
, ,
lim lim
1 x 1 x
i i
= =
|
|
.
|
\
|
=
P T f
P T f
x P T f
P T f
P T
L
i
L
i
i
L
i
L
i
i
x
x (600)
Cos come il coefficiente di fugacit viene correlato allenergia libera di Gibbs residua
parziale molare dalla relazione (567), il coefficiente di attivit viene correlato allenergia
100
Si noti che per quelle grandezze molari il cui valore di miscela ideale coincide con la somma delle
grandezze dei composti puri per la frazione molare (come lentalpia o il volume), le grandezze di
eccesso coincidono con quelle di miscelazione. Ne consegue che, per esempio, il volume molare di
eccesso coincide col volume molare di miscelazione e pu essere quindi misurato sperimentalmente.
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
189
libera di Gibbs di eccesso parziale molare integrando la relazione (395) tra una condizione
di miscela non ideale e una di miscela ideale:
( )
( )
|
|
.
|
\
|
=
=
=
x
x
x x
x
x
x
x
, ,
, ,
ln
) , , ( ) , , (
ln
, ,
, , ,
, ,
, , ,
P T f
P T f
RT
P T P T
f d RT d
i
i
i i
P T
P T
i
P T
P T
i
(601)
Ricordando che il potenziale chimico di un composto in miscela coincide con lenergia
libera di Gibbs parziale molare e utilizzando la definizione di funzione di eccesso, la
relazione precedente pu essere riscritta come:
) , , ( ln ) , , ( ) , , ( ) , , ( x x x x P T RT P T G P T G P T G
i
E
i i i
= =
(602)
Analogamente a quanto fatto per il caso delle funzioni residue, la relazione fondamentale
per una miscela omogenea lequazione (237):
= =
+ + = + + =
N
i
i i
N
i
i i
dn G VdP SdT dn VdP SdT dG
1 1
(603)
Questa relazione consente il calcolo della variazione dellenergia libera di Gibbs sulla base
della variazione di temperatura, pressione e numero di moli.
Esattamente come fatto per la trattazione delle funzioni residue, inserendo la relazione
precedente e la definizione di G=H-TS nellidentit:
dT
RT
G
dG
RT RT
G
d
2
1
= |
.
|
\
|
(604)
si ottiene:
=
+ = |
.
|
\
|
N
i
i
i
dn
RT
G
dT
RT
H
dP
RT
V
RT
G
d
1
2
(605)
Il valore di eccesso della funzione d(G/RT) viene dedotto facilmente:
= =
=
+ =
|
|
.
|
\
|
+
+ + =
=
|
|
.
|
\
|
|
.
|
\
|
=
|
|
.
|
\
|
N
i
i
E
i
E E N
i
i
i
N
i
i
i
E
dn
RT
G
dT
RT
H
dP
RT
V
dn
RT
G
dT
RT
H
dP
RT
V
dn
RT
G
dT
RT
H
dP
RT
V
RT
G
d
RT
G
d
RT
G
d
1
2
1
2
1
2
(606)
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
190
Inserendo la relazione (602) si ottiene infine:
=
+ =
|
|
.
|
\
|
N
i
i i
E E E
dn dT
RT
H
dP
RT
V
RT
G
d
1
2
ln
(607)
Come per le funzioni residue, dalla funzione G
E
/RT possono essere calcolati i valori di
eccesso delle altre funzioni termodinamiche:
( )
n , T
E E
P
RT G
RT
V
(
(
=
(608)
( )
n , P
E E
T
RT G
T
RT
H
(
(
=
(609)
( )
i j
n P T
i
E
i
n
RT G
(
(
=
, ,
ln
(610)
RT
G
RT
H
R
S
E E E
=
(611)
RT
PV
RT
H
RT
U
E E E
=
(612)
Sempre in stretta analogia al caso delle funzioni residue, anche per le funzioni di eccesso si
pone il problema di trovare una relazione con grandezze misurabili.
Questo pu essere fatto utilizzando la relazione (610) riscritta come:
( )
i
E
n P T
i
E
i
RT
G
n
RT G
i j
|
|
.
|
\
|
=
(
(
, ,
ln
(613)
che evidenzia come
i
ln possa essere visto come la grandezza parziale molare di ( ) RT G
E
.
Ne consegue che:
= =
=
|
|
.
|
\
|
=
N
i
i i
N
i
i
E
i
E
n
RT
G
n
RT
G
1 1
ln (614)
o, dividendo entrambi i membri per il numero di moli totale della miscela, in termini
molari:
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
191
=
=
N
i
i i
E
x
RT
g
1
ln
(615)
Questa relazione consente il calcolo dellenergia libera di Gibbs di eccesso (e quindi di tutte
le altre grandezze termodinamiche di eccesso) dai valori dei coefficienti di attivit. Come
detto in precedenza, quando la frazione molare di un composto tende ad uno il suo
coefficiente di attivit tende anchesso ad uno. Poich inoltre ovviamente le frazioni molari
di tutti gli altri composti tendono a zero, deve valere la relazione:
N i x se
RT
g
i
E
... 1 1 0 = = =
(616)
5.6.2.1 Propriet di miscelazione
Dalle relazioni ricavate in precedenza per le diverse propriet di una miscela ideale si pu
ricavare che il volume, lentalpia e lenergia interna molare di miscelazione sono uguali ai
relativi valori di eccesso. Infatti:
E
N
i
i i
mix
h P T h P T h P T h x P T h h = = =
=
) , , ( ) , , ( ) , ( ) , , (
1
x x x (617)
E
N
i
i i
mix
u P T u P T u P T u x P T u u = = =
=
) , , ( ) , , ( ) , ( ) , , (
1
x x x (618)
E
N
i
i i
mix
v P T v P T v P T v x P T v v = = =
=
) , , ( ) , , ( ) , ( ) , , (
1
x x x (619)
Di queste propriet il volume e lentalpia di miscelazione molare sono quelle di maggior
interesse in quanto possono essere misurate sperimentalmente.
Infatti, miscelando due composti puri a temperatura e pressione costante, la differenza tra il
volume finale della miscela e quello iniziale dei due composti rappresenta il volume di
miscelazione, mentre il calore scambiato a pressione costante per mantenere costante la
temperatura pari allentalpia di miscelazione.
5.6.2.2 Misura dei coefficienti di attivit
Rimane al momento irrisolto il problema di misurare i coefficienti di attivit. Questo pu
essere fatto attraverso misure P v T dei composti puri che costituiscono la miscela (o, in
termini equivalenti, utilizzando unopportuna EoS per tali fluidi puri), e misure P v T -
x della miscela in condizioni di equilibrio liquido - vapore. Considerando un recipiente che
contiene una miscela separata in una fase liquida e in una fase vapore in condizioni di
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
192
equilibrio, possibile misurare la temperatura, la pressione, e la composizione di entrambe
le fasi
101
.
Quando in un sistema multicomponente sono presenti pi di una fase, le condizioni di
equilibrio termico e meccanico discusse nella sezione 1.4 richiedono che la temperatura e la
pressione siano uguali in tutte le fasi. Inoltre, affinch il sistema sia in equilibrio anche
rispetto al trasferimento di materia tra le fasi necessario che il potenziale chimico di tutti i
composti nelle diverse fasi assuma lo stesso valore. Considerando per esempio un sistema
contenente una fase liquida e una fase vapore in equilibrio deve essere soddisfatta la
seguente relazione per tutte le specie presenti:
( ) ( ) y x , , , , P T P T
V
i
L
i
= (620)
dove, con una notazione che verr conservata nel prossimo capitolo, si indicato con x la
composizione della fase liquida e con y quella della fase vapore.
Il potenziale chimico ha un legame immediato con la fugacit sulla base della relazione
(561), che integrata tra le condizioni delle due fasi in equilibrio diventa:
|
|
.
|
\
|
= =
L
i
V
i L
i
V
i
P T V
P T L
i
P T V
P T L
i
f
f
RT P T P T f d RT d
ln ) , , ( ) , , (
ln
, , ,
, , ,
, , ,
, , ,
x y
y
x
y
x
(621)
Inserendo la relazione (376) si ricava la seguente condizione, in tutto equivalente
alluguaglianza dei potenziali chimici, che rappresenta la condizione di equilibrio al
trasferimento di materia tra le due fasi:
) , , (
) , , (
x y P T f P T f
L
i
V
i
= (622)
In altri termini, affinch il sistema sia in equilibrio anche rispetto al trasferimento di materia
tra le fasi necessario che la fugacit di tutti i composti nelle diverse fasi assuma lo stesso
valore.
Introducendo in questultima relazione la definizione di coefficiente di attivit (599), si
ottiene:
i
L
i
V
i
i
x P T f
P T f
P T
) , (
) , , (
) , , (
y
x = (623)
Note da misure sperimentali T, P e y possibile calcolare, utilizzando unopportuna EoS, la
fugacit del composto nella miscela in fase vapore. Analogamente, nota la tensione di
vapore possibile calcolare la fugacit del composto puro in fase liquida con il metodo
101
Dovrebbe essere ben noto dai corsi di chimica di base che due fasi in equilibrio hanno solitamente
una diversa composizione. Si accetti comunque per il momento questo come un dato di fatto che verr
ampiamente discusso nel capitolo seguente.
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
193
indiretto. Nota da misure sperimentali anche la composizione della fase liquida, la relazione
(623) consente il calcolo dei coefficienti di attivit da grandezze misurabili.
Si noti che se la fase vapore si comporta come un gas perfetto e la correzione di Poynting
trascurabile la relazione precedente pu essere approssimata dalla seguente equazione:
i i
i
i
x T P
Py
P T
) (
) , , (
0
= x
(624)
5.6.2.3 Modelli di coefficienti di attivit
I coefficienti di attivit dipendono da temperatura, pressione e composizione. Mentre la
dipendenza dalla pressione solitamente debole, quella dalla temperatura e dalla
composizione molto pi marcata
102
.
Sono stati sviluppati diversi modelli per rappresentare la dipendenza dei coefficienti di
attivit dalla composizione, alcuni essenzialmente empirici, altri con fondamenti teorici.
Considerando il semplice caso di una miscela con due componenti (in cui quindi il vincolo
stechiometrico impone x
2
= 1 - x
1
), un semplice modello in grado di rispettare il vincolo
dato dalla (616) il seguente
103
:
) (
2 1
x F x x
RT
g
E
=
(625)
dove F(x) si indica una funzione generica della composizione. Una forma della F(x) molto
flessibile la seguente, nota come espansione di Redlich - Kister
104
:
( ) ( ) ... ) (
2
2 1 2 1
2 1
+ + + = = x x D x x C B
RT x x
g
F
E
x (626)
Considerando un diverso numero di termini nella relazione precedente si ottengono diversi
modelli.
Se tutti i termini sono nulli (B = C = D = = 0) si ottiene la relazione:
102
Questo un caso particolare della regola generale secondo cui le propriet delle fasi condensate
dipendono poco, per valori medio bassi della pressione, dalla pressione. Come ordine di grandezza,
in condizioni non estreme la riduzione di 1 [K] della temperatura ha lo stesso effetto sui coefficienti
di attivit dellaumento della pressione di 50 [bar].
103
Quelli che seguono sono casi semplici di una trattazione generale basata su funzioni razionali, in
cui cio g
E
vene espressa come rapporti di polinomi. Si veda per esempio H.C. Van Ness e M.M.
Abbott, Classical thermodynamics of nonelecrolyte solutions, McGraw-Hill, N.Y., 1982.
104
O. Redlich, A.T. Kister, C.E. Turnquist, Chem. Eng. Progr. Symp. Ser. No. 2, 48, 49-61, 1952.
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
194
0
2 1
=
RT x x
g
E
(627)
da cui, utilizzando la relazione (613) si pu ricavare il valore del coefficiente di attivit:
( ) ( )
0 ln
, , , ,
=
(
(
=
(
(
=
i j i j
n P T
i
E
n P T
i
E
i
n
RT ng
n
RT G
(628)
o, in altri termini, 1 =
i
. Si tratta quindi ovviamente di una miscela ideale.
Se solo il primo termine diverso da zero (C = D = = 0) si ottiene la relazione:
B
RT x x
g
E
=
2 1
(629)
e quindi
( ) ( )
( )
( )
( )
2
2 1 2
2 1
1
2 1
2
2
2 1
2
1
2 1
2
, ,
2 1
2 1
1
, ,
2 1
1
, ,
1
2 1
, ,
1
2 1
, ,
1
1
1 1
ln
2
2 2
2
2
Bx x Bx
n n
n
n n
n
B
n n
n
n
n n
n
B
n n
n n
n
B
n
n
n
n
n
n
B
n
x nBx
n
RT x nBRTx
n
RT ng
n P T
n P T n P T
n P T
n P T
E
= =
|
|
.
|
\
|
+
+
=
=
|
|
.
|
\
|
+
+
=
(
(
|
|
.
|
\
|
+
=
=
(
|
|
.
|
\
|
=
(
=
=
(
=
(
(
=
(630)
Analogamente, si ricava:
2
1 2
ln Bx = (631)
Questa relazione nota come modello di Margules
105
a un parametro
106
. Landamento delle
diverse funzioni previste da questo modello in funzione di x
1
sono riportate in Figura 24 per
B = 1. Si nota che g
E
/(x
1
x
2
RT) ovviamente costante, mentre g
E
/RT simmetrica rispetto a
x
1
= 0,5 e gli andamenti dei due coefficienti di attivit sono speculari. Il parametro B pu
105
Max Margules, 1856 1920, meteorologo e fisico austriaco.
106
Lespansione di Redlich - Kister pu essere trasformata in una forma polinomiale alternativa ed
equivalente (anche se meno immediata) nota come espansione di Margules.
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
195
assumere valori sia positivi sia negativi ed il suo valore viene stimato per confronto coi dati
misurati sperimentalmente.
Figura 24 Andamento delle diverse funzioni secondo il modello di Margules a un
parametro con B = 1.
Se B > 0 ne consegue che anche g
E
e
i
ln sono positivi. In questo caso si dice che il
sistema mostra una deviazione positiva dal comportamento di miscela ideale.
Analogamente, se B < 0 anche g
E
e
i
ln sono negativi e si dice che il sistema mostra una
deviazione negativa dal comportamento di miscela ideale. I valori dei coefficienti di attivit
a diluizione infinita
107
,
i
ln , risultano entrambi uguali a B:
107
Con diluizione infinita si intende una miscela in cui la frazione molare del composto tende a zero.
Si noti, per inciso, che la funzione g
E
/(x
1
x
2
RT) rimane finita anche per x
1
= 0 o x
2
= 0. In particolare,
risulta che
=
1
2 1
0
ln lim
1
RT x x
g
E
x
e
=
2
2 1
0
ln lim
2
RT x x
g
E
x
. Si lascia allo studente la dimostrazione
utilizzando la regola de lHpital e lequazione di Gibbs - Duhem.
0 0.1 0.2 0.3 0.4 0.5 0.6 0.7 0.8 0.9 1
0
0.2
0.4
0.6
0.8
1
x
1
g
E
/(x
1
x
2
RT)
ln
1
ln
2
g
E
/(RT)
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
196
( ) ( ) ( ) B Bx
j
x
i
x
i
x
i
j j i
= = = =
2
1 1 0
lim ln lim ln lim ln
(632)
Se i primi due termini sono diversi da zero (D = = 0) si ottiene la relazione:
( )
2 12 1 21 2 1
2 1
x A x A x x C B
RT x x
g
E
+ = + = (633)
dove A
12
= B C e A
21
= B + C. In questo caso g
E
/(x
1
x
2
RT) non pi costante, ma risulta
lineare in x
1
. Da questa relazione, con passaggi analoghi a quelli visti in precedenza, si
ricava:
( ) ( )
( ) ( )
2 12 21 12
2
1 2
1 12 21 12
2
2 1
2 ln
2 ln
x A A A x
x A A A x
+ =
+ =
(634)
Queste relazioni sono note come modello di Margules a due parametri.
Il valore dei coefficienti di attivit a diluizione infinita risulta pari a:
21 2
12 1
ln
ln
A
A
=
=
(635)
che rappresentano i valori di g
E
/(x
1
x
2
RT) per x
1
= 0 e x
2
= 0, rispettivamente. Il modello di
Margules a due parametri solitamente utilizzato per sistemi simmetrici, cio tali per cui
A
12
A
21
. Se A
12
= A
21
il modello di Margules a due parametri degenera nel modello di
Margules a un parametro.
Unaltra equazione molto utilizzata si deriva assumendo che il reciproco della funzione
(626) sia lineare in x
1
:
( )
'
12
2
'
21
1
2 12 1 21 2 1
2 1
A
x
A
x
x A x A x x C B
g
RT x x
E
+ = + = + =
(636)
dove A
12
= B C e A
21
= B + C. Da questa relazione, con passaggi analoghi a quelli visti
in precedenza, si ricava:
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
197
2
1
'
12
2
'
21
'
21
2
2
2
'
21
1
'
12
'
12
1
1
ln
1
ln
|
|
.
|
\
|
+
=
|
|
.
|
\
|
+
=
x A
x A
A
x A
x A
A
(637)
Queste relazioni sono note come modello di van Laar
108
.
Il valore dei coefficienti di attivit a diluizione infinita per questo modello risultano pari a:
'
21 2
'
12 1
ln
ln
A
A
=
=
(638)
che rappresentano i valori di g
E
/(x
1
x
2
RT) per x
1
o x
2
= 0, rispettivamente. Il modello di van
Laar solitamente utilizzato per sistemi non simmetrici in cui 2
'
21
'
12
A A , mentre non
in grado di rappresentare sistemi in cui
i
ln presenti un minimo o un massimo in funzione
di x
i
.
Questi modelli sono in grado di correlare con successo i dati sperimentali relativi a miscele
a due componenti, ma a causa dei loro inadeguati fondamenti teorici
109
i tentativi di
estenderli a miscele con pi di due componenti o per diverse temperature risultano non
sempre efficaci.
Le teorie pi moderne si basano sul concetto di composizione locale, definita come una
composizione differente da quella media della miscela a causa dellorientamento non
casuale delle molecole come risultato delle diverse dimensioni e interazioni tra le differenti
molecole. Questo concetto ha portato allo sviluppo di diversi modelli, quali il capostipite
modello di Wilson
110
, il modello NRTL
111
(acronimo di Non Random Two Liquids), il
modello UNIQUAC
112
(acronimo di UNIversal QUAsi Chemical)
113
.
108
Johannes Jacobus van Laar, 1860 1938, chimico fisico tedesco
109
Qui si sono presentati questi modelli come puramente correlativi, dedotti cio da una forma
funzionale ipotizzata. Essi possono per anche essere giustificati teoricamente sulla base della teoria
di Wohl (K. Wohl, Trans. Am. Inst. Chem. Engrs., 42, 215, 1946).
110
G.M. Wilson, J. Am. Chem. Soc., 86, 127, 1964.
111
H. Renon e J.M. Prausnitz, AIChE J., 14, 135, 1968.
112
D.S. Abrams e J.M. Prausnitz, AIChE J., 21, 116, 1975.
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
198
La discussione di queste teorie esula dagli scopi di questo testo. Qui basta riportare le
relazioni finali del modello di Wilson per una miscela con un numero arbitrario di
componenti:
=
=
=
= =
|
.
|
\
|
=
|
.
|
\
|
=
N
k N
j
kj j
ki k
N
j
ij j i
N
i
N
j
ij j i
E
x
x
x
x x
RT
g
1
1
1
1 1
ln 1 ln
ln
(639)
in cui 1 =
ii
e
ij ji
. Si noti che queste relazioni contengono, anche per una miscela
con N componenti, solo parametri binari, relativi cio a coppie di composti. Tali parametri
possono essere stimati per confronto con dati sperimentali relativi a miscele di due
composti. La previsione dei coefficienti di attivit di una miscela a N composti richiede
quindi la conoscenza di dati sperimentali relativi solamente a tutte le miscele a due
componenti che si possono formare accoppiando gli N composti. Questo rende affrontabile
in pratica il problema del calcolo dei coefficienti di attivit di miscele contenenti molti
composti.
La dipendenza dei parametri del modello di Wilson dalla temperatura data, in forma
approssimata, dalla seguente relazione:
|
|
.
|
\
|
RT
a
v
v
ij
j
i
ij
exp (640)
dove v il volume molare del composto puro, mentre a rappresenta una costante
indipendente dalla temperatura e dalla composizione.
Il modello di Wilson non in grado di rappresentare sistemi in cui
i
ln presenti un
minimo o un massimo o sistemi in cui si abbia smiscelazione
114
. Il modello NRTL non
presenta particolari vantaggi per miscele moderatamente non ideali, ma in grado di
prevedere adeguatamente il comportamento di miscele fortemente non ideali o parzialmente
113
anche possibile predire il valore dei parametri del modello UNIQUAC utilizzando un
approccio tipo contributi di gruppo, basato cio solo sulla conoscenza dei gruppi funzionali
che costituiscono le molecole dei composti presenti nella miscela (tale approccio viene
chiamato UNIFAC, acronimo di UNIquac Functional-group Activity Coefficients; si veda
per maggiori dettagli: A. Fredenslund, J. Gmehling, P. Rasmussen, Vapor-liquid
equilibrium using UNIFAC, Elsevier, Amsterdam, 1977). Questo approccio consente di
calcolare i coefficienti di attivit senza disporre di dati sperimentali di miscele.
114
Il problema della miscibilit parziale, e quindi dellequilibrio tra due fasi liquide, verr discusso
nel prossimo capitolo.
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
199
miscibili. Infine, il modello UNIQUAC applicabile anche a miscele di composti con
grandi differenze nelle dimensioni molecolari.
5.6.2.4 Applicazione
Per il sistema benzene (1) / n-eptano (2) e 60 [C] sono stati misurati i seguenti valori dei
coefficienti di attivit
x
1
1
ln
2
ln
0,087 0,2655 0,0047
0,180 0,2462 0,0136
0,404 0,1483 0,0654
0,479 0,1307 0,0831
0,713 0,0498 0,2188
0,907 0,0050 0,4332
Si calcoli il valore dei parametri del modello di Margules a due parametri.
Il valore di g
E
/RT viene facilmente calcolato utilizzando la relazione (615), da cui
immediato calcolare il valore della funzione g
E
/(x
1
x
2
RT), come riportato nella seguente
tabella:
x
1
1
ln
2
ln g
E
/(x
1
x
2
RT)
0,087 0,2655 0,0047 0,3450
0,180 0,2462 0,0136 0,3758
0,404 0,1483 0,0654 0,4107
0,479 0,1307 0,0831 0,4244
0,713 0,0498 0,2188 0,4804
0,907 0,0050 0,4332 0,5314
Landamento della funzione g
E
/(x
1
x
2
RT), in funzione di x
1
abbastanza lineare, come si
vede dalla Figura 25.
La relazione (633) pu essere messa nella forma di un modello lineare nei parametri
115
:
115
Un modello lineare nei parametri ha la forma generale: ( ) ) (
1
x f x
T
Np
i
i i
f y = =
=
. Nel caso in
esame il numero di parametri, Np, pari a 2:
(
=
(
=
1 12 21
12
1
) (
x A A
A
x f
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
200
( ) x x A A A x A x A
RT x x
g
E
2 1 1 12 21 12 2 12 1 21
2 1
+ = + = + = (641)
I parametri possono essere stimati dai dati sperimentali con le poche righe di un codice
MATLAB riportate di seguito e risultano pari a A
12
= 0,327 e A
21
= 0,546. La Figura 25
riporta anche, con la linea continua, i valori della funzione calcolati con questi parametri. Si
pu notare il buon accordo tra i dati sperimentali e quelli calcolati.
%margules stima dei parametri del modello di Margules
% da dati sperimentali di gE/(x1x1RT) vs. x1
% modello nella forma gE/(x1x1RT)=A12+(A21-A12)*x1
%
% R. Rota (2002)
clear all
close all
%valori sperimentali di x1
x=[0.087 0.180 0.404 0.479 0.713 0.907]';
%valori sperimentali di gE/(x1x1RT)
y=[0.3450 0.3758 0.4107 0.4244 0.4804 0.5314]';
F=[ones(size(y)) x]; %matrice F
be=F\y; %stima dei parametri del modello lineare
%calcolo di A12 e A21 dai parametri del modello lineare
A12=be(1)
Il valore dei parametri che rende minima la differenza tra i valori calcolati dal modello, y, e quelli
sperimentali, y
exp
= [0,3450 0,3758 0,4107 0,4244 0,4804 0,5314]
T
, nel senso dei minimi quadrati pu
essere calcolato come:
( )
exp
1
y F F F
T T
=
dove F la matrice delle funzioni calcolate per i diversi valori sperimentali, x = [0,087 0,180 0,404
0,479 0,713 0,907]
T
:
( ) ( )
( ) ( )
(
(
(
=
Ns Np Ns
Np
x f x f
x f x f
...
... ... ...
...
1
1 1 1
F
Ns il numero di dati sperimentali (7 in questo caso). MATLAB consente la stima di tali parametri col
semplice comando:
be = F\y
essendo F la matrice delle funzioni e y il vettore dei dati sperimentali.
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
201
A21=be(2)+be(1)
%visualizzazione su grafico dei risultati
xp=[0 x' 1];
ge=A21*xp+A12*(1-xp);
plot(x,y,'o',xp,ge,'k-')
axis([0 1 0 0.6])
xlabel('x_1')
ylabel('g^E/(x_1x_2RT)')
text(0.01,0.3,'A_{12}=0,327')
text(0.85,0.55,'A_{21}=0,546')
Figura 25 Valori sperimentali (o) e calcolati (-) della funzione g
E
/(x
1
x
2
RT).
5.7 Esercizi
Calcolare il fattore di compressibilit, lentalpia molare residua e lentropia molare residua
di una miscela equimolare di CO
2
e CH
4
a 500 [K] e 500 [bar] con lequazione degli stati
corrispondenti.
Risultato: Z = 1,1; h
R
= -2880 [J mol
-1
] ; s
R
= -5,65 [J mol
-1
K
-1
].
0 0.1 0.2 0.3 0.4 0.5 0.6 0.7 0.8 0.9 1
0
0.1
0.2
0.3
0.4
0.5
x
1
g
E
/
(
x
1
x
2
R
T
)
A
12
=0,327
A
21
=0,546
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
202
Calcolare lentalpia molare residua di una miscela equimolare di CO
2
e CH
4
a 500 [K] e
500 [bar] con lEoS PR.
Risultato: h
R
= -3151 [J mol
-1
].
Calcolare il coefficiente di fugacit dei componenti una miscela equimolare di CO
2
e CH
4
a
500 [K] e 500 [bar] con lEoS PR trascurando i parametri di interazione binaria.
Risultato: 0,84; 1,06.
Calcolare il coefficiente di fugacit dei componenti una miscela equimolare di CO
2
e CH
4
a
500 [K] e 500 [bar] con lEoS del viriale.
Risultato: 0,74; 1,00.
Calcolare il volume molare di una miscela equimolare di CO
2
e CH
4
a 200 [K] e 5000 [bar]
con lEoS RKS.
Risultato: 3,2 10
-5
[m
3
mol
-1
].
Calcolare, con lequazione degli stati corrispondenti, la variazione di entalpia e entropia
molare quando una miscela al 70% di etilene e 30% di propilene a 323 [K] e 10 [bar] viene
portata a 600 [K] e 60 [bar].
Risultato: h
= 4785 [J mol
-1
] ; s
= -3,64 [J mol
-1
K
-1
].
Per una miscela metanolo (1) / nitrometano (2) a 1 [bar] sono stati misurati i seguenti valori
dei coefficienti di attivit
x
1
1
ln
2
ln
0,015 1,0315 0,0063
0,426 0,4196 0,3108
0,747 0,0932 0,8035
0,914 0,0095 1,1876
Si calcoli il valore dei parametri del modello di van Laar.
Risultato: A
12
= 1,487 ; A
21
= 1,408.
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
203
6 Equilibrio tra fasi
Nel capitolo precedente sono state discusse le propriet delle miscele e si discusso come
calcolare la fugacit e quindi il potenziale chimico di un componente di una miscela.
Ricordando che la condizione di equilibrio al trasferimento di materia tra due fasi che il
potenziale chimico di ciascun composto sia lo stesso nelle diverse fasi, le relazioni che
consentono il calcolo della fugacit verranno utilizzate in questo capitolo per caratterizzare
le condizioni di equilibrio tra fasi.
Le fasi in equilibrio possono essere diverse. In questo capitolo si discuteranno i casi
dellequilibrio tra una fase liquida e una vapore (solitamente indicato con lacronimo
anglosassone VLE, Vapor Liquid Equilibrium), tra due fasi liquide (LLE, Liquid Liquid
Equilibrium) e tra una fase liquida e una solida (SLE, Solid Liquid Equilibrium). Per
ciascun caso, dopo una breve descrizione qualitativa dei fenomeni coinvolti si discuteranno
le relazioni generali per la caratterizzazione quantitativa dello stato intensivo del sistema,
cio per il calcolo di tutte le variabili intensive che caratterizzano le fasi in equilibrio.
Infine, verranno discussi alcuni casi limite relativi alle soluzioni diluite, cio a quelle
soluzioni, di rilevante interesse applicativo, in cui la concentrazione di un composto sia
molto piccola.
Al termine dello studio di questo capitolo lo studente dovrebbe conoscere:
la regola delle fasi di Gibbs;
le equazioni che caratterizzano lequilibrio tra due fasi;
il comportamento qualitativo di una miscela liquida in equilibrio con il suo vapore;
il diagramma di fase T P x
1
(o y
1
);
il diagramma di fase isotermo P x
1
(o y
1
) e isobaro T x
1
(o y
1
);
il diagramma di fase x
1
y
1
;
il concetto di azeotropo;
il calcolo del punto di bolla e di rugiada di una miscela coi metodi indiretti;
la legge di Raoult e la legge di Raoult modificata;
il calcolo della fugacit di composti supercritici in miscele liquide;
la costante di Henry;
il calcolo del punto di bolla e di rugiada di una miscela coi metodi diretti;
le condizioni a cui le miscele liquide si smiscelano;
il diagramma di solubilit liquido liquido isobaro T x
1
;
il calcolo delle condizioni di equilibrio liquido liquido a pressione costante;
il diagramma di solubilit vapore liquido liquido isobaro T x
1
per liquidi
immiscibili;
il calcolo delle condizioni di equilibrio vapore liquido liquido a pressione costante;
il diagramma di solubilit solido liquido isobaro T x
1
per solidi immiscibili;
il calcolo delle condizioni di equilibrio liquido solido a pressione costante;
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
204
le leggi limite per sistemi diluiti.
6.1 Regola delle fasi di Gibbs
Nel paragrafo 3.1 si introdotto il concetto di gradi di libert o varianza per una sistema
monocomponente e multifase, definito come il numero di variabili intensive che devono (o
possono) essere assegnate arbitrariamente. Ne consegue che tutte le altre variabili intensive
possono essere calcolate utilizzando i valori assegnati e opportune relazioni
termodinamiche.
Questo concetto pu essere esteso facilmente, utilizzando quando discusso nei capitoli
precedenti, anche a sistemi multicomponente e multifase non reagenti
116
. Considerando un
sistema con F fasi e N composti, il numero delle variabili intensive necessarie a
caratterizzare lo stato intensivo di ciascuna fase (temperatura, pressione e composizione
117
)
pari a 2+(N-1) = N+1. Complessivamente, il numero di variabili intensive che si devono
conoscere per caratterizzare completamente lo stato intensivo di un sistema con F fasi
pari a F(N+1).
Le condizioni di equilibrio, discusse nel paragrafo 1.4, richiedono che la temperatura e la
pressione di ciascuna fase siano uguali, cos come devono essere uguali i valori del
potenziale chimico di tutti i composti nelle diverse fasi. Le relazioni che legano le variabili
intensive del sistema (temperatura, pressione e composizione di ciascuna fase) sono quindi
(nel seguito il pedice indica il composto 1 N, mentre lapice la fase 1 F):
( )relazioni 1
...
...
...
relazioni 1 ...
relazioni 1 ...
1
1
1
1
1
1
= =
= =
= =
= =
F N
F P P
F T T
F
N N
F
F
F
(642)
Complessivamente si possono quindi scrivere (F-1)+(F-1)+N(F-1)=(N+2)(F-1) equazioni
nelle F(N+1) variabili. Perch il sistema di equazioni sia risolubile
118
necessario quindi
116
Col termine non reagenti si intende che le diverse specie si possono ripartire nelle diverse fasi, ma
non si possono avere trasformazioni chimiche. In altri termini, un atomo appartiene sempre e solo alla
stessa molecola.
117
Se la composizione di una fase viene espressa in termini di frazioni molari, il numero di frazioni
molari che si devono assegnare per ciascuna fase pari a N-1 in quanto lN-esimo valore si ricava per
complemento a uno:
=
=
1
1
1
N
i
i N
x x
118
Il numero di variabili F(N+1) sempre uguale o maggiore del numero di equazioni (N+2)(F-1).
Per poter risolvere il sistema perci necessario assegnare il valore di un certo numero di variabili
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
205
assegnare il valore di F(N+1) - (N+2)(F-1) = N-F+2 variabili. Questo il numero di
variabili intensive che devono (o possono) essere assegnate arbitrariamente, cio il
numero di gradi di libert o varianza del sistema:
2 + = F N V (643)
Se per esempio il sistema coinvolge 2 fasi e 2 componenti, bisogna assegnare V = 2
variabili intensive. Ovviamente, se N = 1 la relazione precedente degenera nella relazione
gi discussa per il caso monocomponente: V = 3 F.
Una volta assegnate V variabili intensive, le rimanenti variabili intensive possono essere
calcolate utilizzando i valori assegnati e le relazioni (642). Largomento principale di
questo capitolo proprio come usare le relazioni (642), e in particolare luguaglianza dei
potenziali chimici, per calcolare il valore delle restanti variabili intensive di un sistema in
equilibrio con F fasi e N componenti.
6.2 Condizioni di equilibrio
Come gi discusso per il caso particolare dellequilibrio liquido vapore nella sezione
5.6.2.2, quando in un sistema multicomponente sono presenti pi fasi, le condizioni di
equilibrio discusse nella sezione 1.4 richiedono che la temperatura e la pressione siano
uguali in tutte le fasi. Inoltre, affinch il sistema sia in equilibrio anche rispetto al
trasferimento di materia tra le fasi necessario che il potenziale chimico di tutti i composti
nelle diverse fasi assuma lo stesso valore.
Considerando un sistema contenente due fasi in equilibrio, indicate con e , deve essere
soddisfatta la seguente relazione per tutte le specie presenti:
( ) ( )
x x , , , , P T P T
i i
= (644)
Questa relazione indica una prima caratteristica fondamentale degli equilibri di fase:
solitamente la composizione delle fasi in equilibrio non la stessa, cio
x x .
Il potenziale legato alla fugacit dalla relazione (561), che integrata tra le condizioni delle
due fasi in equilibrio diventa:
=
x
x
x
x
, , ,
, , ,
, , ,
, , ,
ln
P T
P T
i
P T
P T
i
f d RT d (645)
cio:
cos da bilanciare il numero di variabili incognite col numero delle equazioni disponibili. Risolvendo
queste equazioni poi possibile calcolare il valore delle variabili non assegnate.
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
206
|
|
.
|
\
|
=
) , , (
) , , (
ln ) , , ( ) , , (
x
x
x x
P T f
P T f
RT P T P T
i
i
i i
(646)
Inserendo la relazione (644) si ricava la seguente condizione, in tutto equivalente
alluguaglianza dei potenziali chimici, che rappresenta la condizione di equilibrio al
trasferimento di materia tra le fasi:
) , , (
) , , (
x x P T f P T f
i i
= (647)
In altri termini, affinch il sistema sia in equilibrio rispetto al trasferimento di materia tra le
fasi necessario che la fugacit di tutti i composti nelle diverse fasi assuma lo stesso valore.
Dato un sistema formato da N composti che si ripartiscono tra due fasi, le variabili intensive
necessarie a caratterizzare completamente il sistema sono quindi la temperatura e la
pressione (due variabili, considerando implicitamente che le due fasi devono avere la stessa
temperatura e pressione
119
) e le frazioni molari in ciascuna fase (N-1 variabili per ciascuna
fase, ricordando che la somma delle frazioni molari deve essere uguale a 1). In totale, si
hanno quindi 2N variabili intensive. La varianza di questo sistema pari a N, e quindi N
variabili devono essere assegnate per poter calcolare le rimanenti N. Le N equazioni
necessarie per calcolare le N variabili non assegnate sono proprio le relazioni (647), una per
ciascun composto presente nelle due fasi. A queste si aggiungono le due relazioni
stechiometriche per calcolare lN-esima frazione molare nelle due fasi:
1
1
1
1
=
=
=
=
N
i
i
N
i
i
x
x
(648)
Tutti i problemi che verranno discussi nel seguito sono riconducibili alla risoluzione del
sistema di equazioni (647) e (648).
6.3 VLE
Se nel sistema sono presenti due componenti, la varianza pari a V = 4 F. Questo
significa che, poich ci deve essere almeno una fase, al massimo la varianza (cio il numero
di variabili intensive sufficienti a caratterizzare completamente lo stato intensivo del
sistema) assume il valore 3, e quindi lo stato intensivo del sistema pu essere rappresentato
graficamente su di un diagramma tridimensionale T P x
1
(o y
1
). Questo facilita
lillustrazione dei principali comportamenti qualitativi del sistema ed il motivo per cui nel
seguito si discuteranno, a titolo di esempio, sistemi a 2 componenti.
119
Questo verr implicitamente assunto in tutto il capitolo.
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
207
Nelle regioni monofase (tutto liquido o tutto vapore) la varianza pari a 3 e quindi si
devono assegnare tre variabili intensive (tra T, P, x
1
o y
1
120
): un punto nello spazio T P
x
1
(o y
1
) rappresenta quindi uno stato intensivo monofase. Per esempio, il punto F in Figura
26 rappresenta una miscela liquida sottoraffreddata, mentre il punto G una miscela vapore
surriscaldata.
Figura 26 Diagramma di fase T P x
1
(o y
1
) per una miscela a due componenti.
120
La frazione molare dellaltro composto nella stessa fase si calcola immediatamente: x
2
= 1 - x
1
e y
2
= 1 - y
1
. Inoltre, si assegna un solo valore di T e P e non un valore per ciascuna fase in quanto sia la
temperatura sia la pressione delle due fasi in equilibrio devono essere uguali.
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
208
Viceversa, nelle regioni bifase (dove la fase liquida e quella vapore coesistono in
equilibrio) la varianza uguale a 2 e quindi, nello spazio tridimensionale, assegnate due
variabili la terza deve essere fissata. Questo implica che nello spazio T P x
1
(o y
1
) ci
deve essere una superficie che rappresenta il luogo dei punti di equilibrio bifase. Tale
superficie analoga alle linee che separano le regioni di esistenza delle diverse fasi nel
diagramma T P di un fluido puro e consente, una volta fissate due variabili (per esempio
T e x
1
) di identificare il valore della terza variabile (in questo caso, la pressione a cui la
miscela liquida di composizione x
1
alla temperatura T in equilibrio col suo vapore).
Poich sullasse della composizione viene riportata sia la composizione della fase liquida
(x
1
) sia quella della fase vapore (y
1
), esistono due superfici, luna relativa al liquido saturo
che lega le tre variabili T P x
1
in condizioni di equilibrio, e laltra relativa al vapore
saturo che correla le tre variabili T P y
1
in condizioni di equilibrio. Nel diagramma di
Figura 26, la superficie superiore quella del liquido saturo, mentre quella inferiore del
vapore saturo. La regione di spazio compresa tra le due superfici rappresenta la regione
delle miscele bifase: non pu esistere cio nessuna fase in uno stato intensivo caratterizzato
da un punto allinterno di tale regione. Se si miscelano i due composti in proporzioni tali da
ricadere, a una certa temperatura e pressione, allinterno di tale regione essi si ripartiscono,
alla stessa temperatura e pressione, tra una fase liquida e una vapore con composizione
diversa e giacente sulle due superfici del liquido e del vapore saturo, rispettivamente.
I piani verticali a x
1
= 0 e x
1
= 1 rappresentano il composto 2 puro e il composto 1 puro,
rispettivamente. La curva KAC
2
rappresenta la curva della tensione di vapore del composto
2, mentre la curva UBC
1
rappresenta la curva della tensione di vapore del composto 1. I
punti C
1
e C
2
sono i punti critici del composto 1 e 2, rispettivamente. La curva C
1
C
2
rappresenta invece il luogo dei punti critici della miscela. Il punto critico di una miscela,
analogamente a quello di un composto puro, viene definito come il punto in cui le propriet
del liquido e del vapore in equilibrio diventano identiche.
Il fatto che le due superfici del vapore e del liquido saturo siano differenti implica che una
miscela con una data composizione e pressione bolle a una temperatura differente da quella
a cui una miscela vapore con la stessa composizione e pressione condensa. In altri termini,
il punto di bolla (definito come la temperatura o la pressione a cui si forma la prima bolla di
vapore in seno al liquido) e il punto di rugiada (definito come la temperatura o la pressione
a cui si forma la prima goccia di liquido in seno al vapore) solitamente sono diversi. Questa
una differenza importante rispetto al caso dei fluidi puri ed ha numerose importanti
implicazioni pratiche
121
.
Si consideri una miscela liquida sottoraffreddata caratterizzata dal punto F in Figura 26 e si
riduca la pressione in modo isotermo in un sistema chiuso. La composizione ovviamente
non cambia (almeno finch presente una sola fase) e la trasformazione rappresentata da
una linea verticale. Quando la pressione raggiunge il valore del punto L incontra la
superficie del liquido saturo: si forma cio la prima bolla di vapore e P
L
rappresenta la
121
Per esempio, la possibilit di separare i componenti di una miscela attraverso la distillazione.
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
209
pressione di bolla (solitamente indicata con P
b
, dallinglese bubble) di quella miscela a
quella temperatura. La bolla di vapore che si forma, essendo in equilibrio con la miscela
liquida caratterizzata dal punto L, dovr avere la stessa temperatura e pressione (ma
composizione solitamente differente) e dovr giacere sulla superficie del vapore saturo:
sar quindi caratterizzata dal punto V. Le linee che uniscono due fasi in equilibrio (come la
linea LV) vengono chiamate linee conodali (o, con termine anglosassone, tie lines).
Riducendo ancora la pressione fino ad incontrare la superficie del vapore saturo nel punto
W tutta la miscela risulta vaporizzata tranne unultima goccia di liquido: P
W
rappresenta la
pressione di rugiada (solitamente indicata con P
d
, dallinglese dew) di quella miscela a
quella temperatura. Analogamente al punto di bolla, la goccia di liquido, essendo in
equilibrio con la miscela vapore caratterizzata dal punto W, dovr avere la stessa
temperatura e pressione (ma composizione solitamente differente) e dovr giacere sulla
superficie del liquido saturo: sar quindi caratterizzata dal punto Z. Riducendo ancora la
pressione si entra nella regione monofase del vapore surriscaldato.
Questo pu essere meglio visualizzato su un diagramma bidimensionale isobaro o isotermo.
Il diagramma isotermo ottenuto sezionando il diagramma T P x
1
(o y
1
) con un piano
verticale a temperatura costante. Si ottengono diagrammi di fase che possono avere forme
diverse, una delle quali, caratteristica di una miscela ideale, riportata in Figura 27.
Considerando una miscela al 40% del composto 1 ad una pressione di circa 70 [kPa] (punto
F in Figura 27) si nella regione monofase liquida. Abbassando la pressione
isotermicamente il sistema raggiunge la curva del liquido saturo (punto L) e si forma la
prima bolla di vapore. La pressione di bolla di questa miscela liquida alla temperatura
assegnata quindi pari a circa 58 [kPa]. La bolla di vapore che si forma, essendo in
equilibrio con la miscela liquida caratterizzata dal punto L, dovr avere la stessa pressione e
dovr giacere sulla curva del vapore saturo: sar quindi caratterizzata dal punto V ed avr
una frazione molare del composto 1 pari a circa 0,57. Riducendo ancora la pressione fino a
circa 55 [kPa] si arriva al punto M che si trova tra le curve del liquido saturo e del vapore
saturo: non pu quindi esistere una fase in queste condizioni. Il sistema si separa in due fasi
che, essendo in equilibrio, sono collegate da una linea conodale orizzontale passante per il
punto M. la composizione delle due fasi pari a circa 0,5 per il vapore saturo (punto M) e
a circa 0,32 per il liquido saturo (punto M). Riducendo ancora la pressione si arriva ad
incontrare la curva del vapore saturo nel punto W dove tutta la miscela risulta vaporizzata
tranne unultima goccia di liquido. La pressione di rugiada risulta quindi pari a circa 52
[kPa]. Analogamente al punto di bolla, la goccia di liquido, essendo in equilibrio con la
miscela vapore caratterizzata dal punto W, dovr avere la stessa pressione e dovr giacere
sulla curva del liquido saturo: sar quindi caratterizzata dal punto Z con una composizione
pari a circa 0,24. Riducendo ancora la pressione si entra nella regione monofase del vapore
surriscaldato (punto G).
Da questo stesso diagramma anche immediato identificare la tensione di vapore alla
temperatura considerata dei due composti, che risulta pari a circa 42 [kPa] per il composto
2 e a circa 83 [kPa] per il composto 1. Il composto 1 risulta quindi essere il pi volatile.
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
210
Figura 27 Diagramma di fase isotermo P x
1
(o y
1
) per una miscela ideale a due
componenti.
In maniera assolutamente analoga lo stesso percorso di evaporazione di una miscela pu
essere visualizzato su un diagramma bidimensionale isobaro ottenuto sezionando il
diagramma T P x
1
(o y
1
) con un piano orizzontale a pressione costante. Anche in questo
caso si ottengono diagrammi di fase che possono avere forme diverse, una delle quali,
caratteristica di una miscela ideale, riportata in Figura 28. Il significato dei punti indicati
analogo a quello degli stessi punti riportati in Figura 27 e discussi precedentemente.
0 0.1 0.2 0.3 0.4 0.5 0.6 0.7 0.8 0.9 1
40
45
50
55
60
65
70
75
80
85
x
1
, y
1
P
[
k
P
a
]
F
L
W
G
V
Z
liquido
vapore
liquido
saturo
vapore
saturo
M
M'
M''
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
211
Figura 28 Diagramma di fase isobaro T x
1
(o y
1
) per una miscela ideale a due
componenti.
Aumentando la temperatura (per i diagrammi isotermi) o la pressione (per quelli isobari) le
curve di equilibrio traslano man mano verso lalto. Quando viene superata la temperatura
(per i diagrammi isotermi) o la pressione (per quelli isobari) critica del composto 1
122
, le
122
Si assume per semplicit di esposizione che il composto 1 sia caratterizzato da un valore di
temperatura e pressione critica inferiore rispetto al composto 2.
0 0.1 0.2 0.3 0.4 0.5 0.6 0.7 0.8 0.9 1
65
70
75
80
85
90
x
1
, y
1
T
[
C
]
liquido
liquido
saturo
vapore
saturo
vapore
Z
M''
L
M
M'
V
W
G
F
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
212
curve di equilibrio non si possono pi estendere fino al limite x
1
= 1, ma si interrompono
prima come illustrato in Figura 29. Il punto critico della miscela caratterizzato dallavere
tangente orizzontale (punto C in Figura 29) poich le linee conodali, che collegano le due
fasi in equilibrio, sono dei segmenti orizzontali
123
.
Figura 29 Diagramma di fase isotermo P x
1
(o y
1
) a diverse temperature (T
d
> T
C2
> T
b
> T
C1
> T
a
) e isobaro T x
1
(o y
1
) a diverse pressioni (P
d
> P
C2
> P
b
> P
C1
> P
a
) per una
miscela a due componenti. I segmenti orizzontali rappresentano le linee conodali, mentre il
punto C il punto critico. I punti A e B sono analoghi a quelli riportati in Figura 26.
Analogamente, se la temperatura (o la pressione) superiore al valore critico di entrambi i
composti le curve di equilibrio non raggiungono neanche il valore x
1
= 0.
I dati di equilibrio di miscele bicomponente sono spesso rappresentati anche su diagrammi
x y in cui sugli assi sono riportati i valori delle frazioni molari nelle due fasi in equilibrio.
Ciascuna curva corrisponde a un dato valore di pressione o di temperatura, come illustrato
in Figura 30.
123
Come detto in precedenza, il liquido e il vapore in equilibrio devono avere la stessa temperatura e
pressione, e quindi le linee che collegano liquido e vapore in equilibrio devono essere orizzontali su
diagrammi che portano temperatura o pressione in ordinata.
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
213
Figura 30 Diagrammi di fase isobaro x
1
y
1
a diverse pressioni per una miscela ideale a
due componenti.
Il diagramma isotermo riportato in Figura 27 si riferisce a una miscela liquida ideale e a una
miscela gassosa che si comporta come un gas perfetto. Per queste miscele, come verr
discusso pi avanti, la pressione di bolla varia linearmente con la frazione molare e quindi
la curva del liquido saturo su un diagramma P x
1
(o y
1
) risulta essere un segmento di retta
che congiunge i valori della tensione di vapore dei composti puri.
Se il comportamento della miscela liquida risulta non ideale, la curva della pressione di
bolla non pi lineare. Se il sistema mostra delle deviazioni positive dal comportamento di
miscela ideale la curva di bolla si trova al di sopra della relazione lineare, come mostrato
nella Figura 31-A. Quando tale deviazione diventa abbastanza grande rispetto alla
differenza tra le tensioni di vapore dei composti puri, la curva di bolla presenta un massimo,
0 0.1 0.2 0.3 0.4 0.5 0.6 0.7 0.8 0.9 1
0
0.2
0.4
0.6
0.8
1
x
1
y
1
T=5 C
T=75 C
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
214
come mostrato nella Figura 31-B. In questo caso, anche la curva di rugiada presenta un
massimo nello stesso punto. Quindi in questo punto, dove la composizione delle due fasi
risulta essere uguale
124
(cio, nel punto di massimo x
1
= y
1
) sia la curva di bolla (P x
1
) sia
quella di rugiada (P y
1
) presentano la stessa tangente orizzontale. Questo punto
chiamato azeotropo.
Figura 31 Diagrammi di fase isotermo P x
1
(o y
1
) per miscele non ideali a due
componenti. La linea tratteggiata rappresenta la curva di bolla di una miscela ideale.
Un discorso assolutamente duale vale nel caso di deviazioni negative dal comportamento di
miscela ideale, come illustrato nella Figura 31-C e D.
124
Un liquido che bolle in questo punto produce quindi un vapore con la stessa composizione, e
quindi il liquido non varia la sua composizione durante levaporazione: non possibile separare i
componenti di una tale miscela per distillazione.
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
215
Deviazioni positive dal comportamento di miscela ideale si riscontrano quando, a livello
molecolare, le forze di attrazione intermolecolari tra molecole uguali sono pi forti di
quelle tra molecole diverse. Ne consegue che lazeotropo risulta essere pi volatile di
entrambi i composti puri. Dualmente, vale unanaloga considerazione per le deviazioni
negative.
Comportamenti analoghi sono riscontrabili anche nei diagrammi T x
1
(o y
1
). Ovviamente,
gli azeotropi di minima pressione appaiono come azeotropi di massima temperatura, e
viceversa.
6.3.1 Calcolo del punto di bolla e di rugiada
Come si discusso in precedenza, in un sistema contenente due fasi e N componenti
necessario assegnare N variabili intensive per poter calcolare le rimanenti N variabili
intensive utilizzando le N relazioni (647). Le relazioni (648) vengono inoltre utilizzate per
calcolare lN-esima frazione molare incognita. Da un punto di vista applicativo
allingegnere si prospetta di solito uno dei seguenti problemi:
dati calcolare nome del problema
T e x P e y calcolo della pressione di bolla
P e x T e y calcolo della temperatura di bolla
T e y P e x calcolo della pressione di rugiada
P e y T e x calcolo della temperatura di rugiada
In altri termini, assegnata la composizione di una fase e la temperatura (o la pressione) si
vuole calcolare la pressione (o la temperatura) a cui quella fase bolle (se liquida) o
condensa (se vapore) e la composizione della prima bolla o goccia dellaltra fase in
equilibrio che si forma.
La soluzione di questo problema passa attraverso la risoluzione, nei casi pi semplici
analitica, altrimenti numerica, del sistema di equazioni algebriche formato dalle relazioni
(647) e da una delle relazioni (648), quella relativa alla fase di cui non si conosce la
composizione. Cos, per il calcolo dei punti di bolla si conosce la composizione della fase
liquida e quindi si utilizzer la 1
1
=
=
N
i
i
y , mentre per il calcolo dei punti di rugiada si
conosce la composizione della fase vapore e quindi si utilizzer la 1
1
=
=
N
i
i
x .
6.3.2 Bassa e media pressione: metodi indiretti (approccio /)
La relazione fondamentale di equilibrio (647) per il caso particolare liquido vapore
diventa:
) , , (
) , , (
x y P T f P T f
L
i
V
i
= (649)
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
216
Esprimendo la fugacit dei composti nella fase vapore con la relazione (564) e quella dei
composti nella fase liquida con la (599), e utilizzando anche la espressione della fugacit di
un composto puro (412), la relazione precedente diviene:
( ) ( ) ( ) ( )
( )
( ) x y , ,
) (
exp , , ,
P T x
RT
T P P v
T P T T P P T Py
i i
o
i
L
i o
i
V
i
o
i
V
i i
|
|
.
|
\
|
= (650)
Questa relazione viene spesso riscritta introducendo il cosiddetto fattore K, definito come il
rapporto tra le frazioni molari nelle due fasi:
( ) ( ) ( )
( )
( )
( ) y
x
, ,
, ,
) (
exp ,
P T P
P T
RT
T P P v
T P T T P
x
y
K
V
i
i
o
i
L
i o
i
V
i
o
i
i
i
i
|
|
.
|
\
|
= =
(651)
I coefficienti di fugacit, tutti relativi alla fase vapore, possono essere calcolati utilizzando
unEoS e i coefficienti di attivit con opportuni modelli di energia libera di Gibbs di
eccesso, come discusso in precedenza.
Questo approccio particolarmente comodo per sistemi a bassa e media pressione. In
questo caso, la correzione di Poynting solitamente trascurabile. Inoltre, il rapporto tra i
coefficienti di fugacit spesso prossimo ad uno:
( )
( ) ( )
1
,
, ,
T P T
P T
o
i
V
i
V
i
y
(652)
cos che la relazione di equilibrio si semplifica nella seguente:
( ) ( ) x , , P T x T P Py
i i
o
i i
(653)
Se poi la miscela liquida ideale, questa relazione si semplifica ulteriormente:
( )
i
o
i i
x T P Py (654)
nota come legge di Raoult
125
. Per questo, la relazione (653) viene spesso indicata come
legge di Raoult modificata.
La legge di Raoult rappresenta il massimo livello di semplificazione della relazione di
equilibrio liquido vapore. Livelli intermedi di semplificazione sono riassunti in Tabella 16.
Analogamente, a seconda delle ipotesi accettabili in un dato caso lespressione del fattore K
si modifica come riassunto in Tabella 17.
125
Franois Marie Raoult, 1830 1901, chimico francese.
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
217
Ipotesi Relazione di equilibrio
Nessuna
( )
( ) ( ) ( )
( )
( ) x
y
, ,
) (
exp ,
, ,
P T x
RT
T P P v
T P T T P
P T Py
i i
o
i
L
i o
i
V
i
o
i
V
i i
|
|
.
|
\
|
=
=
Poynting trascurabile ( ) ( ) ( ) ( ) ( ) x y , , , , ,
P T x T P T T P P T Py
i i
o
i
V
i
o
i
V
i i
=
Poynting trascurabile e
miscela gassosa ideale
( ) ( ) ( ) ( ) ( ) x , , , , P T x T P T T P P T Py
i i
o
i
V
i
o
i
V
i i
=
Poynting trascurabile e gas
perfetto (oppure relazione
(652) valida)
( ) ( ) x , , P T x T P Py
i i
o
i i
=
Poynting trascurabile,
miscela liquida ideale e gas
perfetto (oppure relazione
(652) valida)
( )
i
o
i i
x T P Py =
Tabella 16 Relazioni di equilibrio liquido vapore a diversi livelli di approssimazione.
Ipotesi
Fattore
i i i
x y K =
Nessuna
( ) ( ) ( )
( )
( )
( ) y
x
, ,
, ,
) (
exp ,
P T P
P T
RT
T P P v
T P T T P
V
i
i
o
i
L
i o
i
V
i
o
i
|
|
.
|
\
|
Poynting trascurabile
( ) ( ) ( ) ( )
( ) y
x
, ,
, , ,
P T P
P T T P T T P
V
i
i
o
i
V
i
o
i
Poynting trascurabile e
miscela gassosa ideale
( ) ( ) ( ) ( )
( ) P T P
P T T P T T P
V
i
i
o
i
V
i
o
i
,
, , ,
x
Poynting trascurabile e gas
perfetto (oppure relazione
(652) valida)
( ) ( )
P
P T T P
i
o
i
x , ,
Poynting trascurabile,
miscela liquida ideale e gas
perfetto (oppure relazione
(652) valida)
( )
P
T P
o
i
Tabella 17 Relazioni di equilibrio liquido vapore a diversi livelli di approssimazione.
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
218
6.3.2.1 Punto di bolla
Per il calcolo del punto di bolla si conosce x e quindi si utilizza la 1
1
=
=
N
i
i
y insieme a
una delle relazioni riportate in Tabella 16.
Il caso pi semplice rappresentato dalla legge di Raoult. In questo caso il sistema da
risolvere :
( )
=
=
=
1
1
N
i
i
i
o
i i
y
x T P Py
(655)
e pu essere manipolato per ottenere una sola equazione in una sola incognita, di solito pi
agevole da risolvere rispetto al sistema di N+1 equazioni algebriche (655). Dalla prima
relazione si pu ricavare il valore di y
i:
( )
P
x T P
y
i
o
i
i
=
(656)
che, sostituito nella seconda, porta alla relazione:
( )
0 1
1
=
=
N
i
i
o
i
P
x T P
(657)
Se la temperatura nota si possono calcolare i valori di ) (
0
T P
i
e quindi in modo analitico
la pressione di bolla:
( )
=
=
N
i
i
o
i
x T P P
1
(658)
e la composizione della fase vapore dalla relazione (656).
Se invece nota la pressione, la relazione (657) rappresenta una equazione nellincognita
temperatura che deve essere risolta numericamente
126
. Nota la temperatura, la composizione
della fase vapore pu essere calcolata ancora dalla relazione (656).
Non vi sono sostanziali differenze se si utilizza la legge di Raoult modificata e si trascura,
cosa solitamente lecita, la dipendenza dei coefficienti di attivit dalla pressione. In questo
caso il sistema diventa:
( ) ( )
=
=
=
1
,
1
N
i
i
i i
o
i i
y
T x T P Py x
(659)
126
Per esempio, col comando fzero di MATLAB.
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
219
Dalla prima relazione si ricava il valore di y
i:
( ) ( )
P
T x T P
y
i i
o
i
i
x ,
=
(660)
che, sostituito nella seconda porta alla relazione:
( ) ( )
0 1
,
1
=
=
N
i
i i
o
i
P
T x T P x
(661)
Se la temperatura nota si possono calcolare i valori di ( ) T P
o
i
e di ( ) x , T
i
e quindi la
pressione di bolla risulta dallespressione analitica:
( ) ( )
=
=
N
i
i i
o
i
T x T P P
1
, x (662)
La composizione della fase vapore viene poi calcolata dalla relazione (660).
Se invece nota la pressione, la relazione (661) rappresenta una equazione nellincognita
temperatura che deve essere risolta numericamente. Nota la temperatura, la composizione
della fase vapore pu essere calcolata ancora dalla relazione (660).
Nel caso in cui la correzione di Poynting sia trascurabile e la miscela gassosa ideale i
calcoli sono complicati dalla valutazione del coefficiente di fugacit, che per concerne
solo composti puri e che quindi pu essere facilmente condotta, per pressioni medio - basse,
per esempio con lequazione di stato del viriale. Procedendo in maniera analoga
lequazione risolvente risulta essere:
( ) ( ) ( ) ( )
( )
0 1
,
, ,
1
=
=
N
i V
i
i i
o
i
V o
i
P T P
T x T P T T P
x
(663)
che deve essere risolta numericamente nellincognita pressione o temperatura, a seconda di
quale delle due assegnata. Non possibile ricavare unespressione analitica nemmeno
della pressione, in quanto compare in modo non lineare nellespressione del coefficiente di
fugacit.
Se infine non possibile considerare ideale la miscela gassosa non pi possibile
ricondurre il sistema di equazioni alla risoluzione di una sola equazione algebrica non
lineare in quanto non pi possibile ottenere unespressione analitica di y
i:
Infatti, y
i
compare non linearmente nella espressione del coefficiente di fugacit del composto in
miscela. In questo caso quindi necessario risolvere numericamente il sistema di equazioni
algebriche non lineari
127
:
127
Per esempio, col comando fsolve di MATLAB.
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
220
( )
( ) ( ) ( )
( )
( )
=
=
|
|
.
|
\
|
=
0 1
0 , ,
) (
exp ,
, ,
1
N
i
i
i i
o
i
L
o
i
V o
i
V
i i
y
P T x
RT
T P P v
T P T T P
P T Py
x
y
(664)
6.3.2.2 Punto di rugiada
Per il calcolo del punto di rugiada si conosce y e quindi si utilizza la 1
1
=
=
N
i
i
x insieme a
una delle relazioni riportate in Tabella 16.
Il caso pi semplice rappresentato dalla legge di Raoult. In questo caso il sistema da
risolvere:
( )
=
=
=
1
1
N
i
i
i
o
i i
x
x T P Py
(665)
pu essere manipolato per ottenere una sola equazione in una sola incognita. Dalla prima
relazione si pu ricavare il valore di x
i:
( ) T P
Py
x
o
i
i
i
=
(666)
che, sostituito nella seconda porta alla relazione:
( )
0 1
1
=
=
N
i o
i
i
T P
Py
(667)
Se la temperatura nota si possono calcolare i valori di ( ) T P
o
i
e quindi in modo analitico
la pressione di bolla:
( )
=
=
N
i
o
i i
T P y
P
1
1
(668)
e la composizione della fase liquida dalla relazione (666).
Se invece nota la pressione, la relazione (667) rappresenta una equazione nellincognita
temperatura che deve essere risolta numericamente.
Se invece si utilizza anche solo la legge di Raoult modificata, nasce il problema che i
coefficienti di attivit dipendono in modo non lineare dalla composizione della fase liquida
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
221
e quindi non pi possibile ottenere unespressione analitica di x
i:
In questo caso quindi
necessario risolvere numericamente il sistema di equazioni algebriche non lineari:
( ) ( )
=
=
=
0 1
0 , ,
1
N
i
i
i i
o
i i
x
P T x T P Py x
(669)
nelle incognite x e T oppure P.
Assolutamente analogo il caso delle altre relazioni di equilibrio. Anchesse non possono
essere ricondotte alla soluzione di una sola equazione, ma richiedono sempre la soluzione
numerica di un sistema di equazioni algebriche non lineari. Nel caso pi complicato tale
sistema :
( ) ( ) ( ) ( )
( )
( )
=
=
|
|
.
|
\
|
=
0 1
0 , ,
) (
exp , , ,
1
N
i
i
i i
o
i
L
o
i
V o
i
V
i i
x
P T x
RT
T P P v
T P T T P P T Py x y
(670)
6.3.2.3 Applicazione
Lequilibrio liquido vapore di una miscela cloroformio (1) - metanolo (2) pu essere
rappresentato, a pressione non troppo elevata, dalla legge di Raoult modificata. Si calcolino
la pressione di bolla e di rugiada di una miscela al 40% di cloroformio a 53,5 [C]. Per la
stessa miscela si calcoli la temperatura di bolla e di rugiada a 1 [bar]. I parametri di van
Laar e di Antoine nella forma: log
10
P = A B/(T +C) [torr], T in [C], sono riportati nella
seguente tabella. Si trascuri la dipendenza dei coefficienti di attivit dalla temperatura e
pressione.
composto A B C A
12
A
21
cloroformio (1) 6.95465 1170.966 226.232
metanolo (2) 8.08097 1582.271 239.726
0.9726 1.9210
A 53,5 [C] le tensioni di vapore dei composti puri valgono rispettivamente, per il
composto 1 e 2, 587 e 484 [torr], mentre per x
1
= 0,4 i coefficienti di attivit valgono 1,72 e
1,13 per il composto 1 e 2.
La pressione di bolla risulta quindi pari a:
( ) ( ) [torr] 732 13 , 1 6 , 0 484 72 , 1 4 , 0 587
1
= + = =
=
N
i
i i
o
i b
x T P P x (671)
La composizione della fase vapore in equilibrio si calcola come:
( ) ( )
55 , 0
760
72 , 1 4 , 0 587
1 1 1
1
=
= =
P
x T P
y
o
x
(672)
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
222
Ripetendo questi conti per diverse composizione della fase liquida, per esempio utilizzando
il programma in linguaggio MATLAB riportato di seguito (come al solito commentato e
quindi di facile lettura e che pu essere implementato in un file testo di nome bd.m)
possibile costruire lintero grafico isotermo riportato in Figura 32.
Figura 32 Diagramma di fase isotermo P x
1
(o y
1
) per il sistema cloroformio (1)
metanolo (2) a 53,5 [C]. La curva rappresenta i valori calcolati con un calcolo del punto
di bolla, mentre i simboli quelli calcolati con un calcolo del punto di rugiada.
%bd calcolo punto di bolla e rugiada di
% mix non ideale con Raoult modificata
%
% R. Rota (2002)
0 0.1 0.2 0.3 0.4 0.5 0.6 0.7 0.8 0.9 1
60
65
70
75
80
85
90
95
100
x
1
, y
1
P
[
k
P
a
]
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
223
clear all
close all
format short
global AntA AntB AntC x P gam
%sistema cloroformio(1)/metanolo(2) Rao pag 434
%parametri di Antoine: T [C] e P [torr]; log(Po)=A-B/(T+C)
AntA =[6.95456 8.08097];
AntB =[1170.966 1582.271];
AntC =[226.232 239.726];
%parametri di van Laar
A=0.9726;
B=1.9210;
%P bolla
T =53.5 %[C]
x =[0:0.05:1]; %frazione molare liq. 1
Po=10.^(AntA-AntB./(T+AntC)); %tensione di vapore
gam(1,:)=exp(A./(1+A*x./(B*(1-x))).^2); %coeff. di attivita'
gam(2,:)=exp(B./(1+B*(1-x)./(A*x)).^2);
P =Po(1)*x.*gam(1,:) + Po(2)*(1-x).*gam(2,:); %P di bolla
K1=Po(1).*gam(1,:)./P; %fattore K
y =K1.*x; %frazioni molari di 1 in fase vapore
x(9),P(9),y(9) %visualizzo risultati
figure(1)
plot(x,P./7.6,'k-',y,P./7.6,'k-')
axis('square')
xlabel('x_1, y_1')
ylabel('P [kPa]')
pause
La temperatura di bolla richiede invece la soluzione numerica di unequazione algebrica
non lineare, come illustrato nel programma in linguaggio MATLAB riportato di seguito (che
rappresenta la continuazione del programma bd.m riportato sopra). Si noti come, sfruttando
le propriet vettoriali di MATLAB, possibile costruire lintero grafico isotermo con un solo
comando di risoluzione di un sistema di equazioni algebriche, una per ciascun valore di
composizione considerato. Il programma principale richiede una funzione per il calcolo
della funzione da azzerare, implementata nel file sumxP.m
I risultati ottenuti sono riassunti nel diagramma isobaro riportato in Figura 33. Per una
frazione molare in fase liquida di 0,4 si ottiene una temperatura di bolla di 54,5 [C] e una
composizione della fase vapore pari a 0,55.
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
224
Figura 33 Diagramma di fase isobaro T x
1
(o y
1
) per il sistema cloroformio (1)
metanolo (2) a 760 [torr]. La curva rappresenta i valori calcolati con un calcolo del punto
di bolla, mentre i simboli quelli calcolati con un calcolo del punto di rugiada.
%T bolla
P =760 %[torr]
Teb=-(AntC+AntB./(log10(P)-AntA)); %T bolla dei puri a P, [C]
To =Teb(2)+x.*(Teb(1)-Teb(2)); %valore primo tentativo lineare
T =fsolve('sumxP',To); %T di bolla [C]
Po=10.^(AntA(1)-AntB(1)./(T+AntC(1))); %tensione di vapore
K1=Po.*gam(1,:)./P; %fattore K
y =K1.*x; %frazioni molari di 1 in fase vapore
0 0.1 0.2 0.3 0.4 0.5 0.6 0.7 0.8 0.9 1
50
55
60
65
x
1
, y
1
T
[
C
]
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
225
x(9),T(9),y(9) %visualizzo risultati
figure(2)
plot(x,T,'k-',y,T,'k-')
axis('square')
xlabel('x_1, y_1')
ylabel('T [C]')
pause
function F=sumxP(T)
%SumxP calcola la funzione da azzerare
% per il calcolo della T di bolla
%
% R. Rota (2002)
global AntA AntB AntC x P gam
F=10.^(AntA(1)-AntB(1)./(T+AntC(1))).*x.*gam(1,:) ...
+ 10.^(AntA(2)-AntB(2)./(T+AntC(2))).*(1-x).*gam(2,:)-P;
Il calcolo del punto di rugiada richiede sempre la soluzione di un sistema di equazioni non
lineari, come illustrato nel programma in linguaggio MATLAB riportato di seguito (che
rappresenta sempre la continuazione del programma bd.m riportato sopra e richiede due
funzioni esterne per il calcolo delle funzioni da azzerare, Py.m e Ty.m). Anche in questo
caso il programma consente di calcolare facilmente gli interi diagrammi isobari e isotermi,
che sono riportati coi simboli nella Figura 32 e Figura 33. Si vede come ovviamente i
calcoli di bolla o di rugiada portino agli stessi risultati. In particolare, per una frazione
molare in fase vapore di 0,4 si ottiene una pressione di rugiada di 655 [torr] e una
composizione della fase liquida pari a 0,21, e una temperatura di rugiada di 57,2 [C] e una
composizione della fase liquida pari a 0,22.
%P rugiada
global A B Y Po
yv =[0:0.05:1]; %frazione molare vap. 1
T =53.5 %[C]
Po=10.^(AntA-AntB./(T+AntC)); %tensione di vapore
for j=1:length(y)
Y =yv(j);
Pp=Po(2)+Y.*(Po(1)-Po(2)); %valore primo tentativo lineare
X =fsolve('Py',[Pp Y]); %calcolo P e x1
P(j)=X(1); %memorizzo risultati
xv(j)=X(2);
end
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
226
xv(9),P(9),yv(9) %visualizzo risultati
figure(1)
hold on
plot(xv,P./7.6,'ko',yv,P./7.6,'ko')
axis('square')
xlabel('x_1, y_1')
ylabel('P [kPa]')
pause
%T rugiada
global A B Y P AntA AntB AntC
P =760 %[torr]
for j=1:length(y)
Y =yv(j);
Teb=-(AntC+AntB./(log10(P)-AntA)); %T bolla puri a P, [C]
To =Teb(2)+x.*(Teb(1)-Teb(2)); %primo tentativo ,ineare
X =fsolve('Ty',[To Y]); %calcolo T e x1
T(j)=X(1); %memorizzo risultati
xv(j)=X(2);
end
xv(9),T(9),yv(9) %visualizzo risultati
figure(2)
hold on
plot(xv,T,'ko',yv,T,'ko')
axis('square')
xlabel('x_1, y_1')
ylabel('T [C]')
axis([0 1 50 65])
function F=Py(X)
%Py calcola la funzione da azzerare
% per il calcolo della P di rugiada
%
% R. Rota (2002)
global A B Y Po
P=X(1);
y=[Y 1-Y];
x=[X(2) 1-X(2)];
gam(1)=exp(A/(1+A*x(1)/(B*x(2)))^2);
gam(2)=exp(B/(1+B*x(2)/(A*x(1)))^2);
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
227
F=P.*y - Po.*x.*gam;
function F=Ty(X)
%Ty calcola la funzione da azzerare
% per il calcolo della T di rugiada
%
% R. Rota (2002)
global A B Y P AntA AntB AntC
T=X(1);
y=[Y 1-Y];
x=[X(2) 1-X(2)];
gam(1)=exp(A/(1+A*x(1)/(B*x(2)))^2);
gam(2)=exp(B/(1+B*x(2)/(A*x(1)))^2);
Po=10.^(AntA-AntB./(T+AntC));
F=P.*y - Po.*x.*gam;
6.3.2.4 Composti supercritici
La relazione generale (650) richiede la conoscenza della tensione di vapore del composto
puro alla temperatura della miscela. Se uno dei componenti la miscela presenta una
temperatura critica inferiore a quella della miscela non ovviamente possibile calcolare la
sua tensione di vapore alla temperatura della miscela
128
e lapproccio descritto non pu pi
essere utilizzato. Questo il caso comune dei gas disciolti nei liquidi, per esempio ossigeno
in acqua a condizioni ambiente.
Landamento generale della fugacit di un composto in miscela liquida pu essere misurato
sperimentalmente da misure di equilibrio liquido vapore sulla base della relazione
generale
) , , (
) , , (
x y P T f P T f
L
i
V
i
= (673)
Note da misure sperimentali T, P, x e y possibile calcolare, utilizzando unopportuna EoS,
la fugacit del composto nella miscela in fase vapore, che uguale a quella in fase liquida.
Landamento generale della fugacit in fase liquida in funzione della composizione della
stessa fase riportato in Figura 34.
128
La tensione di vapore richiesta per calcolare, con un approccio indiretto, la fugacit del composto
puro in fase liquida alla temperatura e pressione della miscela.
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
228
Figura 34 Andamento della fugacit di un composto in una miscela liquida in funzione
della composizione. Le linee tratteggiate indicano la legge di Lewis - Randall e di Henry.
Lintercetta in x
1
= 1 ovviamente uguale alla fugacit del composto puro in fase liquida
alla stessa temperatura e pressione della miscela. Inoltre, la curva della fugacit tangente
in x
1
= 1 alla legge lineare di Lewis - Randall per le miscele ideali
129
:
129
Pu essere dimostrato utilizzando la relazione di Gibbs Duhem che
i
x
i
i
f
dx
f d
i
=
|
|
.
|
\
|
=1
0 0.1 0.2 0.3 0.4 0.5 0.6 0.7 0.8 0.9 1
0
200
400
600
800
1000
1200
1400
1600
x
1
f
i
[
k
P
a
]
Lewis / Randall
Henry
H
f
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
229
( )
( )
( )
( )
( )
( ) ( ) P T f
x
x
P T f
x
P T f
P T f
x
x P T
P T f
x
P T f
L
i
i
i
x
L
i
i
L
i
x
L
i
i
i i
x
L
i
i
L
i
x
i i
i i
, lim ,
, ,
lim
,
, ,
lim ,
, ,
lim
1
,
1
1 1
=
|
|
.
|
\
|
=
|
|
.
|
\
|
=
|
|
.
|
\
|
=
|
|
.
|
\
|
x
x x
(674)
Se il composto 1 presenta una temperatura critica inferiore a quella della miscela, non
ovviamente possibile misurare la sua fugacit nella miscela liquida nellintero intervallo di
composizione tra 0 e 1 poich esso non pu esistere come liquido puro alla temperatura
della miscela. In altri termini, la curva riportata in Figura 34 parte da x
1
= 0 (in cui anche la
fugacit del composto 1 in miscela uguale a zero) ma termina prima di x
1
= 1. Questo
significa che linformazione sperimentale ( ) P T f
L
i
, non disponibile, e deve quindi essere
sostituita da unaltra informazione sperimentalmente disponibile. Poich, come si detto,
solamente la parte di sinistra della curva riportata in Figura 34 sperimentalmente
accessibile per composti supercritici, possibile misurare la pendenza di tale curva
allorigine, che viene solitamente chiamata costante di Henry
130
, H
i
:
( )
( )
i j i
x
i
L
i
i
L
i
x
x P T H
dx
f d
x
P T f
i
i
=
=
|
|
.
|
\
|
=
|
|
.
|
\
|
, ,
, ,
lim
0
0
x
(675)
Sia la fugacit sia la frazione molare tendono a zero quando la frazione molare tende a zero,
e quindi largomento del limite diviene indeterminato. Lapplicazione della regola de
lHpital mostra che il limite uguale alla tangente della curva allorigine.
Le relazioni (674) e (675) forniscono due leggi limite, entrambi lineari, per il calcolo della
fugacit di un composto in miscela quando la frazione molare del composto stesso tende a
zero o a uno:
( )
( )
( )
=
1 se ,
0 se , ,
, ,
i i
L
i
i i i j i L
i
x x P T f
x x x P T H
P T f x (676)
La costante di Henry dipende dalla temperatura (molto) e dalla pressione (poco, come tutte
le propriet delle fasi condensate), ma non dalla frazione molare del composto 1 che
prossima a zero. Dipende invece ovviamente dalla composizione della miscela in cui il
composto supercritico si discioglie, cio dal valore delle frazioni molari degli altri
componenti la miscela
131
.
130
Joseph Henry, 1797 1878, fisico statunitense.
131
In altri termini, la costante di Henry dellossigeno in acqua a temperatura e pressione ambiente
diversa da quella dellossigeno in toluene alle stesse condizioni.
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
230
Il problema come sostituire questa nuova informazione sperimentale a quella della
tensione di vapore
132
. Per definizione di coefficiente di attivit si ha che:
( ) ( )
( )
( )
( )
( )
( )
( ) P T f
x P T H
x
P T f
P T f
x P T f
P T f
P T
L
i
i j i
i
L
i
x
L
i
i
L
i
L
i
x
i i
x
i
i i
,
, ,
, ,
lim
,
1
,
, ,
lim , , lim
0
0 0
=
|
|
.
|
\
|
=
=
|
|
.
|
\
|
= =
x
x
x
(677)
da cui si ricava
133
:
( )
=
i
i L
i
H
P T f
,
(678)
La fugacit del composto supercritico in fase liquida si pu quindi scrivere come:
( ) ( ) ( )
( )
( )
( )
( )
( )
( ) P T
P T
x x P T H
P T x
P T
x P T H
P T x P T f P T f
i
i
i i j i
i i
i
i j i
i i
L
i
L
i
,
, ,
, ,
, ,
,
, ,
, , , , ,
=
= = =
x
x x x
(679)
Il rapporto ( ) ( ) P T P T
i i
, , ,
x pu essere definito come un nuovo coefficiente di attivit:
( )
( )
( ) x
x
, ,
,
, ,
P T
P T
P T
i
i
i
(680)
per cui si dice vale la convenzione asimmetrica. Con questo si intende che mentre per i
coefficienti di attivit utilizzati fino a questo momento vale una convenzione simmetrica,
cio il coefficiente di attivit tende a 1 quando anche la frazione molare tende a 1:
( ) ( ) 1 , , lim
1
=
x P T
i
x
i
(681)
per il coefficiente di attivit definito dalla relazione (680) vero il contrario, cio esso
tende a 1 quando la frazione molare tende a 0:
132
In altri termini, si tratta di trovare una relazione che consenta di calcolare il valore di f
i
(T,P)
utilizzando la costante di Henry invece della tensione di vapore.
133
Si noti che il valore di pressione per cui vale questa relazione quello che si ha quando la frazione
molare del composto 1 va a zero. Per una miscela a due componenti si tratta della tensione di vapore
del composto 2 puro. Poich per la correzione di Poynting solitamente trascurabile per pressioni
medio basse, questo valore di fugacit non varia sensibilmente con la pressione.
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
231
( ) ( )
( )
( )
( )
( )
1
,
,
,
, ,
lim , , lim
0 0
= =
|
|
.
|
\
|
=
P T
P T
P T
P T
P T
i
i
i
i
x
i
x
i i
x
x (682)
Quindi, per una miscela contenente sia composti supercritici (cio con una temperatura
critica inferiore a quella della miscela) sia subcritici (cio con una temperatura critica
superiore a quella della miscela), le relazioni di equilibrio liquido vapore sono, per i
composti subcritici:
( ) ( ) ( ) ( )
( )
( ) x y , ,
) (
exp , , ,
P T x
RT
T P P v
T P T T P P T Py
i i
o
i
L
o
i
V o
i
V
i i
|
|
.
|
\
|
=
(683)
mentre per quelli supercritici:
( ) ( )
( )
( ) P T
P T
x x P T H P T Py
i
i
i i j i
V
i i
,
, ,
, , , ,
x
y
(684)
Il calcolo del punto di bolla e di rugiada per tali miscele si svolge esattamente come
descritto in precedenza, con lunica accortezza di utilizzare le relazioni (684) per i composti
supercritici e le (683) per quelli subcritici.
6.3.3 Alta pressione: metodi diretti (approccio /)
La relazione di equilibrio liquido vapore (649) pu essere anche espressa sfruttando la
capacit di alcune equazioni di stato di rappresentare il comportamento sia della fase vapore
sia di quella liquida come:
( ) ( ) x y , ,
, ,
P T Px P T Py
L
i i
V
i i
= (685)
Semplificando il valore della pressione si ottiene la relazione:
( ) ( ) x y , ,
, ,
P T x P T y
L
i i
V
i i
= (686)
o, in termini di fattore K:
( )
( ) y
x
, ,
, ,
P T
P T
x
y
K
V
i
L
i
i
i
i
= = (687)
I coefficienti di fugacit dei composti sia nella fase liquida sia in quella vapore possono
essere calcolati utilizzando unopportuna EoS, per esempio di tipo cubico. Il principale
vantaggio di questo approccio che le equazioni di stato sono intrinsecamente capaci di
tener conto dellinfluenza della temperatura e della pressione. Ovviamente, questo
approccio pu anche essere utilizzato per medie basse pressioni, ma sconta la limitazione
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
232
di un deciso incremento della complessit di calcolo. Infatti, per una data EoS la risoluzione
del sistema di N equazioni algebriche (685) pi una delle relazioni stechiometriche richiede
anche la ricerca delle radici della cubica per il calcolo dei coefficienti di fugacit.
La soluzione del sistema di equazioni pu essere ottenuta con diversi metodi numerici. A
titolo di esemplificazione, lo schema logico per il calcolo della pressione di bolla attraverso
un metodo iterativo
134
il seguente:
sono dati il valore di T e x;
si assume un valore di primo tentativo di P e y (le incognite del problema);
si risolve la cubica per la miscela coi valori di T, P e x; se ci sono tre soluzioni reali
distinte e positive si prende il valore minore di Z, poich si sta analizzando la fase
liquida; con questo valore di Z si calcola il valore di ( ) x , ,
P T
L
i
per tutti i composti;
si risolve la cubica per la miscela coi valori di T, P e y; se si trovano tre soluzioni reali
distinte e positive si prende il valore maggiore di Z, poich si sta analizzando la fase
vapore; con questo valore di Z si calcola il valore di ( ) y , ,
P T
V
i
per tutti i composti;
si calcola il valore di ( ) ( ) y x , ,
, ,
P T P T K
V
i
L
i i
= per tutti i composti;
si calcola il valore di
= =
= =
N
i
i i
N
i
i
x K y S
1 1
;
si ottiene una nuova stima di S x K y
i i i
= e con questi nuovi valori di y si risolve la
cubica per la miscela; se si trovano tre soluzioni reali distinte e positive si prende il
valore maggiore di Z, poich si sta analizzando la fase vapore; con questo valore di Z
si calcola il nuovo valore di ( ) y , ,
P T
V
i
per tutti i composti; si calcola poi il nuovo
valore di ( ) ( ) y x , ,
, ,
P T P T K
V
i
L
i i
= per tutti i composti e quindi il nuovo valore di
= =
= =
N
i
i i
N
i
i
x K y S
1 1
; se il nuovo valore di S uguale al precedente, questo
significa che i valori di y utilizzati per il nuovo calcolo sono uguali a quelli utilizzati
per il vecchio calcolo e che quindi le equazioni (687), che sono quelle utilizzate per
calcolare y, sono soddisfatte;
a questo punto bisogna verificare se anche la equazione stechiometrica soddisfatta; se
S = 1 i valori di P e y utilizzati sono corretti e il calcolo terminato; altrimenti si
sceglie un diverso valore di P = P S e si ripete il procedimento dal punto 3 fino a
convergenza.
Viene lasciato allo studente lo sviluppo di procedure analoghe per il calcolo della pressione
di rugiada e della temperatura di bolla e di rugiada.
134
Nellapplicazione seguente si illustrer luso del comando MATLAB fsolve.
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
233
6.3.4 Applicazione
Si calcoli la pressione di bolla di una miscela equimolare di propilene (1) e n-butano (2) a
300 [K] utilizzando lEoS PR.
Lapplicazione della procedura iterativa presentata sopra la seguente:
T = 300 [K] e x = [0,5 0,5], dai dati del problema;
P = 7,173 [bar] e y = [0,8232 0,1768], calcolati utilizzando la legge di Raoult;
si risolve, come illustrato nel capitolo precedente, la cubica per la miscela coi valori di
T, P e x e si trovano le tre soluzioni Z = [0,02526 0,12168 0,83526]. Si utilizza quindi
il valore di Z = 0,02526, poich si sta analizzando la fase liquida; con questo valore di
Z si calcola, come illustrato nel capitolo precedente, il valore di
( ) 0,34123] 1,3319 [ , ,
= x P T
L
per i due composti;
analogamente, si risolve la cubica per la miscela coi valori di T, P e y e si trovano le
soluzioni Z = [0,023782 0,089716 0,87066]. Si prende questa volta il valore maggiore,
Z = 0,87066, poich si sta analizzando la fase vapore; con questo valore di Z si calcola
il valore di ( ) 0,82244] 0,89817 [ , ,
= x P T
V
per i due composti;
si calcola il valore di ( ) ( ) y x , ,
, ,
P T P T K
V
i
L
i i
= per i due composti e si ottiene K =
[1,4829 0,4149];
si calcola il valore di 0,9489
1 1
= = =
= =
N
i
i i
N
i
i
x K y S ;
si ottiene una nuova stima di S x K y
i i i
= e con questi nuovi valori di y = [0,78138
0,21862] si risolve nuovamente la cubica per la miscela, trovando i valori di Z =
[0.02396 0,093493 0,86646], di cui si considera il valore Z = 0,86646 poich si sta
analizzando la fase vapore; con questo valore di Z si calcola il nuovo valore di
( ) 0,34327] 1,3277 [ , ,
= y P T
V
per i due composti; si calcola poi il nuovo valore di
( ) ( ) y x , ,
, ,
P T P T K
V
i
L
i i
= per i due composti (K = [1,4782 0,41738]) e quindi il
nuovo valore di 9478 , 0
1 1
= = =
= =
N
i
i i
N
i
i
x K y S ; il nuovo valore di S non
sufficientemente simile al precedente e quindi si devono ripetere i conti del punto 7
finch due valori di S calcolati in due passi successivi risultano sufficientemente simili;
a questo punto bisogna verificare se anche la equazione stechiometrica soddisfatta; se
S = 1 i valori di P e y utilizzati sono corretti e il calcolo terminato; altrimenti si
sceglie un diverso valore di P = P S e si ripete il procedimento dal punto 3 fino a
convergenza.
Al termini delle iterazioni si ottiene il risultato: P = 6,743 [bar] e y = [0,7822 0,2178].
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
234
Lo stesso risultato pu essere ottenuto con molta minor fatica utilizzando un solutore di
sistemi algebrici non lineari come quello disponibile in MATLAB col comando fsolve.
Questo mostrato nel listato, al solito commentato e quindi di facile comprensione, del
programma in linguaggio MATLAB riportato di seguito che pu essere implementato in un
file testo di nome VLE_mix.m).
%VLE_mix calcolo della P di bolla
% di una miscela a T, y con diverse EoS
% per miscele binarie
%
% R. Rota (2002)
format short g %formato scrittura
format compact %formato scrittura
clear all %cancella tutte le variabili
global T x RT RTc TR Tc Pc om
%dati per propilene
Tc(1) =365.1; %[K]
Pc(1) =46.00e5; %[Pa]
om(1) =0.181; %fattore acentrico di Pitzer
Zc(1) =0.274; %fattore di compressibilita' critico
%dati per n-butano
Tc(2) =425.2; %[K]
Pc(2) =37.97e5; %[Pa]
om(2) =0.199; %fattore acentrico di Pitzer
Zc(2) =0.274; %fattore di compressibilita' critico
%assegno le condizioni di T e x1
T = 300; %temperatura [K]
x(1)= 0.5; %frazione molare del composto 1
x(2)= 1-x(1); %frazione molare del composto 2
%valori di primo tentativo secondo Raoult
Po(1) = exp(15.7027-1807.53/(T-26.15))/760; %P [atm]
Po(2) = exp(15.6782-2154.90/(T-34.42))/760;
Pp =sum(Po.*x);
yp =Po(1)*x(1)/Pp;
%azzero il sistema di equazioni phiLi xi = phiVi yi
%nelle incognite P e y1
Xo(1) =Pp; %P primo tentativo [bar]
Xo(2) =yp; %y1 primo tentativo
var = fsolve('phiVL',Xo)
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
235
Il codice richiama la funzione phiVL, implementata in un file di testo di nome phiVL.m e
in cui vengono calcolate le equazioni da risolvere.
function F = phiVL(X)
%F calcola phiLi*xi phiVi*yi in miscela binaria
% con EoS RK, RKS o PR
%
% R. Rota (2002)
global T x Tc Pc om
P = X(1)*1e5; %P in [Pa]
y(1) = X(2);
y(2) = 1-X(2);
%calcolo A e B per i composti puri
tipo=4; %EoS PR
for j=1:2,
[Z,Ap(j),Bp(j),S,k] = EoS(T,P,Tc(j),Pc(j),om(j),tipo);
end
%ciclo su gas (fase=1) e liquido (fase=2)
for fase = 1:2;
if fase == 1
%calcolo Z a (P,T,y)
[Z,A,B,STrk] = EoS_mix(T,P,y,Tc,Pc,om,tipo);
phi(fase,:)=exp(lnphi(Ap,Bp,A,B,Z(3),tipo)).*y; %Zvap
else
[Z,A,B,STrk] = EoS_mix(T,P,x,Tc,Pc,om,tipo); %calcolo Z
a (P,T,x)
phi(fase,:)=exp(lnphi(Ap,Bp,A,B,Z(1),tipo)).*x; %Zliq
end
end
F = diff(phi);
Questo codice utilizza i programmi EoS, EoS_mix e lnphi, gi discussi nelle applicazioni
precedenti. Risulta evidente come, sfruttando i programmi gi sviluppati, sia possibile con
pochi comandi effettuare i tediosi calcoli iterativi illustrati in precedenza.
Modificando semplicemente lassegnazione della composizione della fase liquida poi
possibile ripetere i calcoli per diverse composizioni
135
, per diverse temperature e con
135
immediato costruire il diagramma di equilibrio liquido vapore con un metodo diretto
aggiungendo solo pochi comandi al programma VLE_mix. Questo sviluppo viene lasciato allo
studente per esercizio.
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
236
diverse equazioni di stato. A titolo di esempio, la tabella seguente riporta i risultati dei conti
con due diverse EoS.
PR RKS
P
b
[bar] 6,74 6,81
y
1
0,78 0,78
6.4 LLE
In condizioni di equilibrio a temperatura e pressione costante lenergia libera di Gibbs,
come discusso nella sezione 4.2, deve assumere il minimo valore compatibile coi vincoli
imposti. Quindi, la miscelazione di due o pi composti a temperatura e pressione costante
risulta essere un processo spontaneo solo se accompagnato da una diminuzione
dellenergia libera di Gibbs. In altri termini, se lenergia libera di Gibbs della miscela
risulta essere minore della somma di quella dei composti puri questi si miscelano
spontaneamente; se viceversa lenergia libera di Gibbs della miscela risulta essere maggiore
di quella risultante dalla somma dellenergia libera di Gibbs dei composti puri essi non si
miscelano spontaneamente e risultano quindi essere completamente immiscibili. Esistono
ovviamente anche delle situazioni intermedie per cui i composti sono solo parzialmente
miscibili
136
: cio possibile miscelarli in certe proporzioni, ma non in altre. In altri termini,
la miscela pu esistere solo entro certi limiti di composizione.
Questo pu essere illustrato considerando lenergia libera di una miscela che, per una mole
di una miscela a due componenti, pu essere espressa dalla relazione:
( ) ) , , ( ln ln ) , ( ) , (
) , , ( ) , , ( ) , , (
2 2 1 1 2 2 1 1
x
x x x
P T g x x x x RT x P T g x P T g
P T g P T g P T g
E
E
+ + + + =
= + =
(688)
Utilizzando per esempio il modello di Margules a un parametro:
RT x Bx g
E
2 1
= (689)
possibile calcolare lenergia libera di Gibbs di miscele a diversa composizione al variare
del valore del parametro B. La Figura 35 mostra landamento dellenergia libera di Gibbs di
una miscela binaria calcolato con le relazioni sopra al variare del parametro B
137
.
136
Risulta essere unesperienza comune che acqua e olio non si miscelano completamente nonostante
siano entrambi liquidi nelle condizioni ambiente. Si forma cio una fase acquosa contenente un po di
olio disciolto e una fase oleosa contenente un po di acqua disciolta.
137
Si assunto a titolo di esempio g
1
/RT = 0,5 e g
2
/RT = 0; le conclusioni che si traggono da questa
figura sono ovviamente generali.
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
237
Figura 35 Andamento dellenergia libera di Gibbs con la composizione di una miscela
binaria al variare del parametro B del modello di Margules a un parametro.
Per B = 0 (cio per miscela ideale) lenergia libera della miscela sempre, qualsiasi sia la
composizione, inferiore al valore dellenergia libera di Gibbs dei due composti puri che
pari a g
1
x
1
+g
2
x
2
= (g
1
-g
2
)x
1
+g
2
ed rappresentata dalla retta tratteggiata che congiunge i
valori g
1
e g
2
. Come gi stato discusso, questo significa che la formazione di una miscela
ideale sempre un processo spontaneo.
Per B = 10 (cio per una miscela fortemente non ideale) lenergia libera della miscela
sempre, qualsiasi sia la composizione, maggiore del valore dellenergia libera di Gibbs dei
due composti puri. Questo significa che i due composti sono completamente immiscibili in
0 0.1 0.2 0.3 0.4 0.5 0.6 0.7 0.8 0.9 1
-0.5
0
0.5
1
1.5
2
x
1
g
/
R
T
B=10
B=0
g
2
/RT
g
1
/RT
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
238
quanto, per qualsiasi composizione, la formazione della miscela porterebbe ad un aumento
dellenergia libera di Gibbs del sistema
138
.
Un discorso analogo vale per due miscele distinte di composizione arbitraria, nel senso che
cos come una miscela ideale risulta pi stabile dei composti puri separati, essa risulta
anche pi stabile di due miscele qualsiasi in cui pu separarsi. Con riferimento alla Figura
36 si considerino due miscele di composizione
1
x e
1
x . Considerando quantit diverse
delle due miscele lenergia libera di Gibbs del sistema complessivo ha un valore compreso
tra
g e
g
139
, e analogamente al caso dei composti puri rappresentata in figura da un
punto sulla linea a tratto e punto che congiunge i due punti e . Viceversa, se le due
miscele vengono mescolate e si miscelano completamente, la miscela risultante deve avere
una composizione compresa tra
1
x e
1
x e la sua energia libera di Gibbs rappresentata da
un punto sulla curva a tratto continuo tra i due punti e . Si vede immediatamente che per
qualsiasi quantit delle due miscele e la somma dellenergia libera delle due miscele
separate sempre superiore a quella della miscela risultante dal loro completo
mescolamento. In altri termini, miscele ideali si mescolano sempre completamente: si dice
che una miscela ideale non pu smiscelarsi.
Esistono ovviamente anche situazioni intermedie, come mostrato nella Figura 37 per il caso
di B = 3.
In questo caso si vede che per un certo intervallo di composizione (nellintervallo tra i punti
A e B in Figura 37) lenergia libera di Gibbs della miscela superiore a quella dei composti
puri separati: in questi intervallo di composizione, come discusso in precedenza, la
formazione della miscela porterebbe ad un aumento dellenergia libera di Gibbs del
sistema.
138
In realt per x
1
0 e x
1
1 esiste un piccolo intervallo di composizione in cui lenergia libera di
Gibbs della miscela inferiore alla somma di quella dei composti puri. Questo significa che in realt i
due composti non sono completamente insolubili, ma che una piccola quantit del composto 1 si
scioglie nel composto 2 e viceversa. Ai fini della presente esemplificazione tale solubilit viene
trascurata.
139
Si continua a considerare una mole complessiva di sostanza; considerando una quantit di materia
diversa nulla cambia se non che nel ragionamento a g si deve sostituire G. Inoltre, in figura lordinata
non g ma bens g/RT, come in precedenza; ovviamente concettualmente non cambia nulla.
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
239
Figura 36 Andamento dellenergia libera di Gibbs con la composizione di una miscela
binaria ideale.
Vi per unimportante differenza rispetto al caso discusso in precedenza considerando
due miscele di composizione
1
x e
1
x . Lenergia libera di Gibbs del sistema complessivo
formato da quantit diverse di queste due miscele separate ha sempre un valore
rappresentato in Figura 37 da un punto sulla linea a tratto e punto che congiunge i due punti
e , cos come se le due miscele vengono mescolate e si miscelano completamente la
miscela risultante ha una composizione compresa tra
1
x e
1
x e la sua energia libera di
Gibbs rappresentata da un punto sulla curva a tratto continuo tra i due punti e .
0 0.2 0.4 0.6 0.8 1
-0.5
-0.3
-0.1
0.1
0.3
0.5
x
1
g
/
R
T
x
1
x
1
/RT
g
/RT
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
240
Figura 37 Andamento dellenergia libera di Gibbs con la composizione di una miscela
binaria per B = 3.
In questo caso, si vede che per qualsiasi quantit delle due miscele e la somma
dellenergia libera delle due miscele separate sempre inferiore a quella della miscela
risultante dal loro completo mescolamento. In altri termini, se si mescolano i due composti
in modo tale da formare una miscela di composizione compresa tra
1
x e
1
x si separano
due fasi liquide distinte in equilibrio: luna di composizione
1
x e laltra di composizione
0 0.1 0.2 0.3 0.4 0.5 0.6 0.7 0.8 0.9 1
-0.1
0
0.1
0.2
0.3
0.4
0.5
x
1
g
/
R
T
x
1
x
1
A
B
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
241
1
x
140
. In questo caso si dice che il sistema parzialmente miscibile, nel senso che per
composizioni comprese tra 0
1
= x e
1 1
x x = , oppure tra
1 1
x x = e 1
1
= x il sistema
completamente miscibile, mentre non lo per composizioni interne allintervallo
] [
1 1
x x
141
.
Pur senza entrare in alcun dettaglio, importante notare che affinch un modello di energia
libera di eccesso di Gibbs sia in grado di prevedere lo smiscelamento di una soluzione
liquida deve essere in grado di riprodurre matematicamente landamento funzionale
esemplificato in Figura 37. Non tutti i modelli sviluppati sono in grado di far questo: per
esempio il modello di Wilson, che non pu quindi essere utilizzato nel caso di LLE.
Lintervallo di composizione entro cui i composti non si miscelano (cio la lacuna di
miscibilit identificata dallintervallo di composizione ] [
1 1
x x in Figura 37) varia con la
temperatura
142
. Analogamente a quanto discusso per il caso di VLE, si possono riassumere
le condizioni di equilibrio tra due fasi liquide per una miscela a due componenti su
diagrammi di fase isobari che riportano la frazione molare di un componente in ascissa e la
temperatura in ordinata
143
, solitamente chiamati diagrammi di solubilit.
Il caso pi generale rappresentato in Figura 38 dove la regione interna alla curva
rappresenta la lacuna di miscibilit. La curva che delimita la lacuna di miscibilit il luogo
140
Questo comportamento del tutto analogo a ci che avviene se si mescolano due composti e la
composizione della miscela risultante compresa tra la composizione della fase liquida e della fase
vapore in equilibrio alla temperatura e pressione assegnata: il sistema si divide in due fasi in
equilibrio, luna liquida e laltra vapore.
141
Analogamente si dice che il sistema presenta una lacuna di miscibilit per composizioni comprese
tra
1
x e
1
x .
142
Ovviamente varia anche con la pressione, ma come tutte le propriet delle fasi condensate tale
variazione non significativa per variazioni modeste della pressione o per valori della temperatura
ridotta abbastanza bassi da poter trascurare leffetto della pressione sui coefficienti di attivit. Nel
seguito verr discusso quindi solo il caso particolare, ma di rilevante importanza pratica, in cui
linfluenza della pressione possa essere trascurata.
143
Come discusso per il caso di VLE, se nel sistema sono presenti due componenti la varianza pari a
4 F e questo significa che lo stato intensivo del sistema pu essere rappresentato graficamente su di
un diagramma tridimensionale T P
1
x (o
1
x ). Fissando la pressione (diagrammi isobari)
possibile rappresentare graficamente lo stato intensivo del sistema su di un diagramma
bidimensionale T
1
x (o
1
x ). Questo facilita lillustrazione dei principali comportamenti qualitativi
del sistema ed il motivo per cui nel seguito si discuteranno, a titolo di esempio, sistemi a 2
componenti.
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
242
dei punti di equilibrio tra le fasi: il tratto di curva UAL rappresentativo della fase liquida
(quella pi ricca del composto 2), mentre il tratto di curva UBL rappresentativo della
fase liquida (quella pi ricca del composto 1).
Figura 38 Diagramma di solubilit liquido liquido isobaro per una miscela a due
componenti che presenta sia LCST sia UCST.
Poich la curva che delimita la lacuna di miscibilit il luogo dei punti di equilibrio tra le
fasi e ricordando che due fasi in equilibrio hanno la stessa temperatura, ne consegue che le
linee conodali su questo diagramma sono segmenti orizzontali. Quindi, se si mescolano i
due composti in proporzioni tali che la composizione della miscela risultante, alla
temperatura e pressione assegnata, interna alla lacuna di miscibilit il sistema si separa in
due fasi distinte, la cui composizione data dallintersezione della retta orizzontale alla
temperatura del sistema coi due rami della curva di equilibrio.
0 0.1 0.2 0.3 0.4 0.5 0.6 0.7 0.8 0.9 1
250
300
350
400
450
x
1
T
[
K
]
UCST
LCST
una fase liquida
due fasi
liquide
x
1
x
1
A
B
linea conodale
curva di equilibrio
U
L
fase liquida fase liquida
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
243
Per esempio, per il sistema rappresentato in Figura 38, alla temperatura di 300 [K] per x
1
<
0,382 e per x
1
> 0,610 si ha una sola fase liquida (o, in altri termini, i due composti sono
completamente miscibili). Per concentrazioni intermedie viceversa il sistema si separa in
due fasi in equilibrio, la cui composizione pari a 382 , 0
1
=
x e 610 , 0
1
=
x . La linea
conodale che unisce i due punti A e B rappresentanti le due fasi liquide e in equilibrio
il tratto di linea orizzontale in Figura 38.
Figura 39 Diagramma di solubilit liquido liquido isobaro per una miscela a due
componenti che presenta solo LCST (destra) o solo UCST (sinistra).
Dalla medesima figura anche evidente leffetto della temperatura sulla composizione delle
due fasi in equilibrio. In particolare si evidenziano due temperature critiche: per valori di
temperatura superiori alla prima (denominata UCST, dallinglese Upper Critical Solution
0 0.2 0.4 0.6 0.8 1
250
300
350
400
450
x
1
T
[
K
]
0 0.2 0.4 0.6 0.8 1
250
300
350
400
450
x
1
T
[
K
]
una fase liquida
una fase liquida
due fasi
liquide
due fasi
liquide
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
244
Temperature) o inferiori alla seconda (detta LCST, dallinglese Lower Critical Solution
Temperature) i due composti sono sempre completamente miscibili
144
.
In realt il comportamento schematizzato in Figura 38 non molto frequente in quanto la
curva di equilibrio tra le due fasi solitamente interseca la curva caratteristica di unaltra
transizione di fase a temperature superiori alla LCST (in questo caso la soluzione solidifica
prima di raggiungere la LCST) o inferiori alla UCST (in questo caso la soluzione bolle
prima di raggiungere la UCST). Questi due comportamenti sono schematizzati nei
diagrammi riportati in Figura 39, la cui interpretazione analoga a quello riportato in
Figura 38.
6.4.1 Calcolo dello stato di equilibrio a media e bassa pressione
In completa analogia con quanto discusso in precedenza per il caso di VLE, in un sistema
contenente due fasi e N componenti necessario assegnare N variabili intensive per poter
calcolare le rimanenti N variabili intensive utilizzando le N relazioni (647). Le relazioni
(648) vengono inoltre utilizzate per calcolare lN-esima frazione molare incognita. La
relazione fondamentale di equilibrio (647) per il caso particolare di equilibrio tra due fasi
liquide diventa:
) , , (
) , , (
x x P T f P T f
L
i
L
i
= (690)
Esprimendo la fugacit dei composti nella fase liquida con la relazione (599) la relazione
precedente diviene:
) , , ( ) , ( ) , , ( ) , (
x x P T x P T f P T x P T f
i i
L
i i i
L
i
= (691)
che si riduce alla:
) , , ( ) , , (
x x P T x P T x
i i i i
= (692)
I coefficienti di attivit che compaiono nella relazione precedente derivano dallo stesso
modello di energia libera di Gibbs di eccesso e si differenziano solo in quanto sono
calcolati a composizioni diverse (quelle della fase e , rispettivamente)
145
.
144
Si noti lanalogia con la temperatura critica incontrata nellequilibrio liquido - vapore. Anche in
questo caso la temperatura critica rappresenta il valore di temperatura limite di un sistema bifase per
cui tutte le propriet delle due fasi diventano uguali.
145
Si noti che se la miscela ideale i coefficienti di attivit sono unitari e quindi le composizioni delle
due miscele sono uguali: in altri termini, non si pu avere smiscelazione.
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
245
6.4.2 Applicazione
Il comportamento del sistema liquido n-esano / furfurolo pu essere rappresentato dal
modello di Margules a un parametro con B = 800/T, T in [K]
146
. Calcolare la mutua
solubilit dei due composti a 298 [K].
La condizione di equilibrio (692) in questo caso pu essere posta in una forma pi utile per
i calcoli come segue:
( ) ( )
( ) ( )
|
|
.
|
\
|
=
|
.
|
\
|
=
|
|
.
|
\
|
|
|
.
|
\
|
=
|
.
|
\
|
=
|
|
.
|
\
|
1
1
2
1
2
1
2
2
1
1
2
1
2
1
1
1
1
1
ln
) , , (
) , , (
ln
ln 1 1
) , , (
) , , (
ln
x
x
x x B
P T
P T
x
x
x x B
P T
P T
x
x
x
x
(693)
che rappresenta un sistema di due equazioni non lineari nelle due incognite
1
x e
1
x e pu
essere risolto numericamente, come mostrato nel listato, al solito commentato e quindi di
facile comprensione, del programma in linguaggio MATLAB riportato di seguito che pu
essere implementato in un file testo di nome LLE.m.
%LLE calcolo dell'equilibrio liq - liq
%
% R. Rota (2002)
clear all
close all
format short
global B
T =298; %temperatura [K]
B =800/T; %calcolo B alla T data
Xo =[0.1 0.9]; %valori di primo tentativo
%risolvo il sistema
var = fsolve(@fLLE,Xo,optimset('TolFun',1e-20))
Il codice richiama la funzione fLLE, implementata in un file di testo di nome fLLE.m e in
cui vengono calcolate le equazioni da risolvere.
146
Si noti che il modello di Margules a un parametro prevede lo smiscelamento solo per B > 2.
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
246
function F = fLLE(X)
%fLLE calcola fialfa - fibeta in miscela binaria
% per LLE nella forma:
% ln(gamma1alfa/gamma1beta)-ln(x1beta/x1alfa) = 0
% ln(gamma2alfa/gamma2beta)-ln(x2beta/x2alfa) = 0
% modello di Margules a un parametro
%
% R. Rota (2002)
global B
x1a=X(1);
x2a=1-X(1);
x1b=X(2);
x2b=1-X(2);
F(1) = B*(x2a^2-x2b^2) - log(x1b/x1a);
F(2) = B*(x1a^2-x1b^2) - log(x2b/x2a);
Il codice fornisce il risultato 109 , 0
1
=
x e 891 , 0
1
=
x , che sono i valori delle
concentrazioni delle due fasi liquide in equilibrio. La mutua solubilit dei due composti
luno nellaltro quindi pari a circa il 10 %.
Con semplici modifiche il programma LLE.m descritto pu essere utilizzato per costruire i
diagrammi di solubilit, quali quelli illustrati in precedenza
147
, come riportato di seguito.
%LLE costruzione dei diagrammi di solubilita
% liq - liq a P = cost
%
% R. Rota (2002)
clear all
close all
format short
global B
147
Per ottenere le tre forme dei diagrammi di solubilit discusse in precedenza il valore del parametro
B deve variare in modo diverso con la temperatura, attraversando comunque il valore B = 2 nel campo
di temperatura di interesse. In particolare, deve essere decrescente con T per sistemi che presentano
solo UCST, crescente con T per sistemi che presentano solo LCST, presentare un massimo e
attraversare due volte il valore B = 2 per sistemi che presentano sia UCST sia LCST.
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
247
% valori dei parametri di B
a =-975;
b =22.4;
c =-3;
xa=[], xb=[], Tp=[]; %inizializzo i vettori ausiliari
Xo =[0.1 0.9]; %valori di primo tentativo
for T=250:0.5:400,
B = a/T+b+c*log(T); %calcolo B alla T data
if B>=2
var = fsolve(@fLLE,Xo,optimset('TolFun',1e-20)); %risolvo il
sistema
Tp = [Tp T]; %memorizzo i risultati
xa = [xa var(1)]; %memorizzo i risultati
xb = [xb var(2)]; %memorizzo i risultati
end
end
plot([xa fliplr(xb) xa(1)],[Tp fliplr(Tp) Tp(1)],'k')
axis('square'), axis([0 1 250 450])
xlabel('x_1'), ylabel('T [K]')
6.4.3 VLLE con composti in fase liquida completamente immiscibili
a bassa e media pressione
Quando la lacuna di miscibilit che rappresenta le condizioni di equilibrio tra due fasi
liquide interseca a una data temperatura la curva di bolla, che rappresenta le condizioni di
equilibrio tra la fase liquida e quella vapore, si ha la simultanea presenza di due fasi liquide
e di una fase vapore (VLLE, dallinglese Vapor Liquid Liquid Equilibrium).
Considerando sempre a titolo esemplificativo un sistema a due componenti, la regola delle
fasi impone che vi sia, in presenza di tre fasi, un solo grado di libert. Quindi, per un valore
di pressione assegnato, risultano fissate sia la temperatura sia la composizione delle tre fasi.
In altri termini, su di un diagramma di solubilit isobaro esiste un solo punto in cui possono
coesistere le tre fasi in equilibrio. Per semplicit di esposizione si considera nel seguito solo
il caso limite, ma di rilevante importanza applicativa, in cui i composti liquidi sono
completamente immiscibili
148
. Il diagramma di solubilit isobaro per una miscela a due
componenti relativo a questa situazione riportato in Figura 40.
In Figura 40 sono visibili i principali comportamenti qualitativi dei sistemi VLLE. Il punto
E (con temperatura pari a TVLLE e composizione pari a x
1
VLLE) rappresenta lunico
148
Si pensi ad esempio al caso dei numerosi sistemi acqua / idrocarburi in cui la mutua solubilit pu
essere spesso trascurata ai fini pratici.
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
248
punto in cui coesistono in equilibrio le due fasi liquide (costituite in questo caso particolare
dai due composti liquidi puri) e la fase vapore (costituita da una miscela dei due composti).
Figura 40 Diagramma di solubilit liquido liquido vapore isobaro per una miscela a
due componenti che presenta completa immiscibilit in fase liquida.
A temperature inferiori a TVLLE esistono solo le due fasi liquide, mentre a temperature
superiori a TVLLE esiste anche la fase vapore. Le due curve rappresentano le curve di
rugiada della fase vapore, mentre la linea orizzontale a TVLLE rappresenta la curva di bolla
delle miscele liquide
149
.
149
TVLLE rappresenta la curva di bolla solo delle miscele. I due composti puri hanno ovviamente
temperatura di bolla e di rugiada coincidenti (nel caso rappresentato in figura circa 383 [K] per il
0 0.1 0.2 0.3 0.4 0.5 0.6 0.7 0.8 0.9 1
340
345
350
355
360
365
370
375
380
385
390
x
1
T
[
K
]
composti 1 e 2 vapore
composti 1 e 2
liquidi immiscibili
TVLLE
x
1
VLLE
E
A
B
C
D
F
I
H
G
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
249
Considerando una miscela con x
1
= 0,8 e T = 375 [K] si ha una miscela in fase vapore
(punto A in Figura 40). Riducendo la temperatura a pressione costante la miscela rimane in
fase vapore fino a quando raggiunge la curva di rugiada (punto B), dove si separa la prima
goccia di liquido. La composizione del liquido che si forma (in completa analogia coi
comportamenti VLE visti in precedenza) in equilibrio con la miscela vapore caratterizzata
dal punto B e dovr quindi avere la stessa temperatura e giacere sulla curva di bolla (le
linee conodali sui diagrammi isobari sono rappresentate da segmenti orizzontali). In questa
regione del diagramma la curva di bolla rappresentata dallasse verticale a x
1
= 1, e quindi
il liquido che si forma costituito dal composto 1 puro.
Riducendo ancora la temperatura fino al punto C che si trova tra le curve di bolla e di
rugiada si entra nella regione bifase, dove cio non pu esistere una singola fase. Il sistema
si separa quindi in due fasi che, essendo in equilibrio, sono collegate da una linea conodale
orizzontale passante per il punto C. La composizione delle due fasi pari a circa 0,69 per il
vapore saturo, mentre il liquido sempre formato dal composto 1 puro. Riducendo ancora
la temperatura si arriva alla TVLLE, dove possono coesistere le tre fasi in equilibrio: quella
vapore con composizione x
1
VLLE e i due composti liquidi puri. Riducendo ancora la
temperatura (punto D) si entra nella regione del liquido sottoraffreddato dove coesistono i
due composti puri immiscibili.
Quando la composizione della miscela vapore iniziale invece che superiore a x
1
VLLE
(punto A) inferiore (punto F , caratteristico di una miscela con x
1
= 0,4), il comportamento
qualitativo della miscela al raffreddamento lo stesso, con la sola differenza che raggiunta
la curva di rugiada si separa il composto liquido 2 puro invece del composto liquido 1 puro.
Il percorso FGHI quindi del tutto analogo a quello ABCD illustrato in precedenza.
Per il caso di VLLE, in un sistema contenente tre fasi e N componenti necessario
assegnare N 1 variabili intensive per poter calcolare le rimanenti 2N variabili intensive.
Le relazioni di equilibrio che si possono scrivere nel caso di VLLE sono le stesse gi viste
per lequilibrio tra una fase vapore e una liquida e tra due fasi liquide:
) , , (
) , , (
) , , (
) , , (
) , , (
) , , (
x x
y x
y x
P T f P T f
P T f P T f
P T f P T f
L
i
L
i
V
i
L
i
V
i
L
i
=
=
=
(694)
composto 2 e 373 [K] per il composto 1). In altri termini, la curva di bolla orizzontale per tutti i
valori di 0
1
x e 1
1
x , per poi diventare verticale e congiungersi con la curva di rugiada per
0
1
= x e 1
1
= x . Si tratta ovviamente di un comportamento limite per composti completamente
immiscibili che, come detto, non esistono anche se molti composti approssimano sufficientemente
bene questo comportamento ai fini pratici; nella realt le due curve (di bolla e di rugiada) convergono
alle temperature di ebollizione dei due composti puri in modo pi graduale e continuo.
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
250
oltre alle relazioni stechiometriche per le tre fasi. Appare evidente che le relazioni (694)
non sono tutte indipendenti: per esempio, le relazioni di equilibrio tra le due fasi liquide
possono essere facilmente dedotte dalle altre. Ne consegue che delle 3N relazioni (694) se
ne possono utilizzare solo 2N che consentono di calcolare le 2N variabili incognite.
Utilizzando quanto visto in precedenza per il caso VLE e LLE si ottiene facilmente,
considerando le relazioni approssimate valide per sistemi a media o bassa pressione
discusse in precedenza:
( ) ( )
( ) ( )
( ) ( )
x x
x
x
, , , ,
, ,
, ,
P T x P T x
Py P T x T P
Py P T x T P
i i i i
i i i
o
i
i i i
o
i
=
=
=
(695)
Nel caso di composti liquidi completamente immiscibili le fasi liquide sono costituite da
specie pure
150
. Per ciascuna delle due fasi liquide si ha quindi che la frazione molare di un
composto unitaria (e quindi anche il suo coefficiente di attivit), mentre quella dellaltro
composto pari a zero. Le relazioni precedenti degenerano quindi nelle seguenti
espressioni:
( )
( )
1 1
2 2
1 1
=
=
=
Py T P
Py T P
o
o
(696)
dove lultima identit riflette il fatto che le frazioni molari dei componenti nelle due fasi
liquide sono unitarie. Le due equazioni non banali consentono di calcolare le due incognite
rimanenti tra le tre variabili intensive necessarie a caratterizzare il sistema (temperatura,
pressione e frazione molare di uno dei due composti nella fase vapore) quando una delle tre
sia fissata. La frazione molare del secondo componente in fase vapore viene calcolata
utilizzando la relazione stechiometrica: y
2
= 1 y
1
.
6.4.3.1 Applicazione
Si calcoli la temperatura di ebollizione del sistema acqua (1) / benzene (2) a 1 [bar]
considerando i due composti completamente immiscibili in fase liquida. La tensione di
vapore in funzione della temperatura pu essere calcolata con lequazione di Antoine
( ) ( ) T C B A P + =
10
log , P in [torr] e T in [K], coi seguenti parametri per lacqua e il
benzene: A = [8,07131 6,87987]; B = [1730,63 1196,76]; C = [-39,5740 -53,8390].
150
Poich si considera la presenza di due fasi si possono quindi avere al massimo due componenti;
stiamo cio considerando il caso particolare di miscele bicomponenti.
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
251
Introducendo le relazioni di equilibrio (696) nella relazione stechiometrica per la fase
vapore si ottiene:
( ) ( )
1
2 1
2 1
=
+
= +
P
T P T P
y y
o o
(697)
Questa rappresenta unequazione nellincognita temperatura attraverso le relazioni di
Antoine:
P
C T
B
A
C T
B
A
= +
|
|
.
|
\
|
+
|
|
.
|
\
|
+
2
2
2
1
1
1
10 10
(698)
che pu essere risolta numericamente, come mostrato nel listato, al solito commentato e
quindi di facile comprensione, del programma in linguaggio MATLAB riportato di seguito
che pu essere implementato in un file testo di nome VLLE.m.
%VLLE calcolo della temperatura di equilibrio VLLE
% per liquidi immiscibili
%
% R. Rota (2002)
clear all
close all
format short
global A B C P
%parametri per le tensioni di vapore
%log10(P) = A-B/(T+C), T in [K] e P in [torr]
%acqua - benzene
A = [8.07131 6.87987];
B = [1730.63 1196.76];
C = [233.426-273 219.161-273];
P = 1; %pressione [bar]
%calcolo della T di VLLE a 1 [bar]
TVLLE = fzero(@fVLLE,340)
Po = 10.^(A - B ./ (TVLLE+C)) ./ 760; %P [bar]
y1 = Po(1)/P
Il codice richiama la funzione fVLLE, implementata in un file di testo di nome fVLLE.m e
in cui viene calcolata lequazione da risolvere.
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
252
function F = fVLLE(T)
%fLLE calcola la T di equilibrio VLLE nella forma:
% P1(T)+P2(T) = P
%
% R. Rota (2002)
global A B C P
Po = 10.^(A-B./(T+C))./760; %tensione di vapore in bar
F = sum(Po) - P;
Il codice fornisce il risultato T = 342,1 [K] e y
1
= 0,296.
Con semplici modifiche il programma VLLE.m descritto pu essere utilizzato per costruire i
diagrammi di equilibrio, quali quelli illustrati in precedenza, come riportato di seguito.
%VLLE diagrammi di solubilit
% vap - liq - liq a P = cost
% per liquidi immiscibili
%
% R. Rota (2002)
clear all
close all
format short
global A B C P
%parametri per le tensioni di vapore
%log10(P) = A-B/(T+C), T in [K] e P in [torr]
%acqua - benzene
A = [8.07131 6.87987];
B = [1730.63 1196.76];
C = [233.426-273 219.161-273];
P = 1; %pressione [bar]
%calcolo della T di VLLE a 1 [bar]
TVLLE = fzero(@fVLLE,340)
Po = 10.^(A - B ./ (TVLLE+C)) ./ 760; %P [bar]
y1 = Po(1)/P
Td=[], xd=[]; %inizializzo le variabili
%calcolo dei due rami del diagramma
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
253
for x=0:.01:1
Pi=[P*x P*(1-x)].*760; %pressioni parziali [torr]
T = B ./ (A-log10(Pi)) - C; %calcolo le T dei due rami
Td= [Td max(T)];
xd= [xd x];
end
plot(xd,Td,'k-',[0 1],[TVLLE TVLLE],'k:')
axis('square'), axis([0 1 330 390])
xlabel('x_1'), ylabel('T [K]')
6.5 SLE
Sistemi che coinvolgono fasi liquide e solide sono normalmente coinvolti nei processi di
cristallizzazione industriale in cui un componente della miscela liquida viene separato come
composto puro in fase solida. A seconda dei diversi possibili comportamenti delle due fasi
(liquida e solida) sono possibili molti comportamenti qualitativi differenti. Nel seguito
verr discusso in dettaglio solo il caso limite, anche se di rilevante importanza applicativa,
di una soluzione liquida priva di lacune di miscibilit e di composti solidi completamente
immiscibili.
Il comportamento qualitativo in tutto simile a quello discusso in precedenza per il caso di
VLLE coi composti in fase liquida completamente immiscibili, pur di sostituire alla fase
vapore la fase liquida e alle due fasi liquide le due fasi solide.
Considerando a titolo esemplificativo un sistema a due componenti, la regola delle fasi
impone che vi sia, in presenza di tre fasi (due solide e una liquida), un solo grado di libert.
Quindi, per un valore di pressione assegnato, risultano fissate sia la temperatura sia la
composizione delle tre fasi e su di un diagramma isobaro esiste quindi un solo punto in cui
possono coesistere le tre fasi in equilibrio. Questo punto viene chiamato eutettico. Il
diagramma isobaro per una miscela a due componenti completamente immiscibili in fase
solida riportato in Figura 41.
In Figura 41 sono visibili i principali comportamenti qualitativi dei sistemi SLE di questo
tipo, che sono analoghi a quelli visti in precedenza per i sistemi VLLE. Il punto E (con
temperatura pari a T
E
e composizione pari a x
1E
) rappresenta lunico punto in cui coesistono
in equilibrio le due fasi solide (costituite in questo caso particolare dai due composti puri) e
la fase liquida (costituita da una miscela dei due composti). A temperature inferiori a T
E
esistono solo le due fasi solide, mentre a temperature superiori a T
E
esiste anche la fase
liquida. Le due curve rappresentano le curve di solidificazione della fase liquida, mentre la
linea orizzontale a T
E
rappresenta la curva di fusione delle fasi solide
151
.
151
T
E
rappresenta la curva di fusione solo dei sistemi bicomponente. I due composti puri hanno
ovviamente temperatura di fusione e di solidificazione coincidenti (nel caso rappresentato in figura
circa 383 [K] per il composto 2 e 373 [K] per il composto 1). In altri termini, la curva di fusione
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
254
Figura 41 Diagramma di solubilit isobaro liquido solido per una miscela a due
componenti che presenta completa immiscibilit in fase solida e completa miscibilit in
fase liquida.
Considerando una miscela con x
1
= 0,8 e T = 375 [K] si ha una miscela in fase liquida
(punto A in Figura 41). Riducendo la temperatura a pressione costante la miscela rimane in
orizzontale per tutti i valori di 0
1
x e 1
1
x , per poi diventare verticale e congiungersi con la
curva di solidificazione per 0
1
= x e 1
1
= x . Si tratta ovviamente di un comportamento limite per
composti completamente immiscibili che, come detto, non esistono anche se molti composti
approssimano sufficientemente bene questo comportamento ai fini pratici; nella realt le due curve
convergono alle temperature di fusione dei due composti puri in modo pi graduale e continuo.
0 0.1 0.2 0.3 0.4 0.5 0.6 0.7 0.8 0.9 1
340
345
350
355
360
365
370
375
380
385
390
x
1
T
[
K
]
composti 1 e 2 liquidi
composti 1 e 2
solidi immiscibili
T
E
x
1E
E
A
B
C
D
F
I
H
G
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
255
fase liquida fino a quando raggiunge la curva di solidificazione (punto B), dove si separa la
prima particella di solido. La composizione del solido che si forma (in completa analogia
coi comportamenti VLLE visti in precedenza) in equilibrio con la miscela liquida
caratterizzata dal punto B e dovr quindi avere la stessa temperatura e giacere sulla curva di
fusione (le linee conodali sui diagrammi isobari sono rappresentate da segmenti
orizzontali). In questa regione del diagramma la curva di fusione rappresentata dallasse
verticale a x
1
= 1 e quindi il solido che si forma costituito dal composto 1 puro.
Riducendo ancora la temperatura fino al punto C che si trova tra le curve di solidificazione
e di fusione si entra nella regione bifase, dove cio non pu esistere una singola fase. Il
sistema si separa quindi in due fasi che, essendo in equilibrio, sono collegate da una linea
conodale orizzontale passante per il punto C. La composizione delle due fasi pari a circa
0,69 per il liquido, mentre il solido sempre formato dal composto 1 puro. Riducendo
ancora la temperatura si arriva alla T
E
, dove possono coesistere le tre fasi in equilibrio:
quella liquida con composizione x
1E
e i due composti solidi puri. Riducendo ancora la
temperatura (punto D) si entra nella regione del solido sottoraffreddato dove coesistono i
due composti puri immiscibili.
Quando la composizione della miscela liquida iniziale invece che superiore a x
1E
(punto A)
inferiore (punto F, caratteristico di una miscela con x
1
= 0,4), il comportamento
qualitativo della miscela al raffreddamento lo stesso, con la sola differenza che raggiunta
la curva di solidificazione si separa il composto solido 2 puro invece del composto solido 1
puro. Il percorso FGHI quindi del tutto analogo a quello ABCD illustrato in precedenza.
Le relazioni di equilibrio che si possono scrivere nel caso di SLE con le fasi solide
immiscibili sono analoghe a quelle gi viste per gli altri equilibri di fase:
) , ( ) , , (
P T f P T f
S
i
L
i
= x (699)
oltre alla relazione stechiometrica per la fase liquida
152
. La fugacit del composto in fase
solida ovviamente relativa al composto puro.
Considerando le relazioni (423) e (623) dalla relazione precedente si ottengono le seguenti
relazioni generali:
( ) ( )
( )
|
|
.
|
\
|
|
|
.
|
\
|
|
|
.
|
\
|
+
|
|
.
|
\
|
=
=
T
T
T
T
R
c c
T T R
h
P T f
P T x P T f
fi fi
S
Pi
L
Pi
fi
fus i L
i
i i
L
i
ln 1
1 1
exp ,
, , ,
,
x
(700)
che forniscono le seguente espressione della solubilit
153
del composto i-esimo:
152
Poich le fasi solide sono costituite da composti puri, le relazioni stechiometriche per queste fasi
degenerano nella uguaglianza banale 1 =
Sj
j
x , essendo j lunico composto presente nella fase solida j.
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
256
( )
|
|
.
|
\
|
|
|
.
|
\
|
|
|
.
|
\
|
+
|
|
.
|
\
|
=
T
T
T
T
R
c c
T T R
h
P T
x
fi fi
S
Pi
L
Pi
fi
fus i
i
i
ln 1
1 1
exp
, ,
1
,
x
(701)
Se poi si considera la forma approssimata della relazione (423) (equazione (424)) si ottiene:
( )
|
|
.
|
\
|
|
|
.
|
\
|
=
T T R
h
P T
x
fi
fus i
i
i
1 1
exp
, ,
1
,
x
(702)
Nel caso di miscela liquida ideale la relazione precedente si semplifica ulteriormente per
dare:
|
|
.
|
\
|
|
|
.
|
\
|
=
T T R
h
x
fi
fus i
i
1 1
exp
,
(703)
Queste relazioni consentono di tracciare le curve di equilibrio dei diagrammi illustrati in
Figura 41. Per calcolare la curva tra 0
1
= x e
E
x x
1 1
= si utilizza la relazione di equilibrio
relativa al composto 2 (in quanto in questa regione vi equilibrio tra la fase liquida e il
composto 2 solido), mentre per la curva tra
E
x x
1 1
= e 1
1
= x si utilizza la relazione di
equilibrio relativa al composto 1.
6.5.1.1 Applicazione
Si tracci il diagramma di solubilit solido liquido del sistema toluene etilbenzene a
pressione atmosferica considerando la miscela liquida ideale. Le propriet del toluene e
delletilbenzene a pressione atmosferica sono pari rispettivamente a: 1580 [cal mol
-1
] e
2168 [cal mol
-1
] per lentalpia di fusione; 178,20 [K] e 178,25 [K] per la temperatura di
fusione.
Nel punto di eutettico devono valere entrambe le relazioni di equilibrio (sia quella per il
toluene composto 1 sia quella per letilbenzene composto 2 ) in quanto vi
equilibrio tra la fase liquida e entrambi i composti solidi:
153
Si definisce in questo caso come solubilit la massima concentrazione di un composto che pu
essere presente disciolto in una fase liquida in condizioni di equilibrio con la propria fase solida pura.
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
257
|
|
.
|
\
|
|
|
.
|
\
|
= =
|
|
.
|
\
|
|
|
.
|
\
|
=
E f
fus
E E
E f
fus
E
T T R
h
x x
T T R
h
x
1 1
exp 1
1 1
exp
2
, 2
1 2
1
, 1
1
(704)
Nella relazione precedente si considerata la relazione di equilibrio approssimata e si
anche utilizzata la relazione stechiometrica per la fase liquida. Da queste relazioni si ottiene
lequazione:
|
|
.
|
\
|
|
|
.
|
\
|
=
|
|
.
|
\
|
|
|
.
|
\
|
E f
fus
E f
fus
T T R
h
T T R
h
1 1
exp 1
1 1
exp
1
, 1
2
, 2
(705)
che rappresenta unequazione nella sola incognita T
E
da risolvere numericamente.
Il diagramma di equilibrio viene poi facilmente costruito utilizzando la relazione di
equilibrio per il composto 1 per valori di temperatura compresi tra T
f1
e T
E
, e quella per il
composto 2 per valori di temperatura compresi tra T
f2
e T
E
. Il problema pu essere risolto
come mostrato nel listato, al solito commentato e quindi di facile comprensione, del
programma in linguaggio MATLAB riportato di seguito che pu essere implementato in un
file testo di nome SLE.m. I risultati dei conti sono riassunti in Figura 42. Lo stesso
programma consente anche il calcolo delle curve di equilibrio con le relazioni di equilibrio
(423), tratteggiate in Figura 42.
%SLE diagrammi di solubilit
% sol - liq a P = cost
% per solidi immiscibili
%
% R. Rota (2002)
clear all
close all
format short
global DHf Tf Dcp
%parametri per toluene (1) - etilbenzene (2)
DHf = [1580 2168]*4.186; %entalpia di fusione [J/mol]
Tf = [178.20 178.25]; %T di fusione [K]
Dcp = [32.4-20.8 37.61-25.32]*4.186; %cpL-cpS [J/mol K]
%calcolo della TE
TE = fzero(@fSLE,160)
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
258
%calcolo curve di solubilita'
%regione in cui si separa il composto 1
T1 = linspace(TE,Tf(1));
x1 = exp(DHf(1)/8.314.*(1/Tf(1)-1./T1)+Dcp(1)/8.314.*(Tf(1)./T1-1-
log(Tf(1)./T1)));
%regione in cui si separa il composto 2
T2 = linspace(Tf(2),TE);
x2 = exp(DHf(2)/8.314.*(1/Tf(2)-1./T2)+Dcp(2)/8.314.*(Tf(2)./T2-1-
log(Tf(2)./T2)));
T = [T2 T1];
x = [1-x2 x1];
plot(x,T,'k:',[0 1],[TE TE],'k:') %calcolo rigoroso
axis('square'), axis([0 1 TE-10 max(Tf)+10]), xlabel('x_1'),
ylabel('T [K]')
hold on
%soluzione approssimata
Dcp = [0 0];
%calcolo della TE
TE = fzero(@fSLE,160)
%calcolo curve di solubilita'
%regione in cui si separa il composto 1
T1 = linspace(TE,Tf(1));
x1 = exp(DHf(1)/8.314.*(1/Tf(1)-1./T1)+Dcp(1)/8.314.*(Tf(1)./T1-1-
log(Tf(1)./T1)));
%regione in cui si separa il composto 2
T2 = linspace(Tf(2),TE);
x2 = exp(DHf(2)/8.314.*(1/Tf(2)-1./T2)+Dcp(2)/8.314.*(Tf(2)./T2-1-
log(Tf(2)./T2)));
T = [T2 T1];
x = [1-x2 x1];
plot(x,T,'k-',[0 1],[TE TE],'k-') %calcolo approssimato
Il codice richiama la funzione fSLE, implementata in un file di testo di nome fSLE.m e in
cui viene calcolata lequazione da risolvere.
function F = fSLE(T)
%fSLE calcola la T di eutettico
%
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
259
% R. Rota (2002)
global DHf Tf Dcp
aux = exp(DHf./8.314.*(1./Tf-1./T)+Dcp./8.314.*(Tf./T-1-
log(Tf./T)));
F = sum(aux) - 1;
Figura 42 Diagramma di solubilit isobaro liquido solido per una miscela toluene -
etilbenzene. La linea continua rappresenta i risultati ottenuti utilizzando la relazione (424),
mentre quella tratteggiata la relazione (423).
0 0.1 0.2 0.3 0.4 0.5 0.6 0.7 0.8 0.9 1
210
220
230
240
250
260
270
280
290
x
1
T
[
K
]
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
260
6.6 Leggi limite per sistemi diluiti
Le relazioni di equilibrio discusse in questo capitolo solitamente si semplificano assumendo
forme molto pi semplici nel caso limite di sistemi diluiti, in cui cio la frazione molare di
un composto tende a zero. Queste forme semplificate delle relazioni di equilibrio vengono a
coincidere con delle leggi o regole dedotte empiricamente prima che la termodinamica
degli equilibri di fase delle miscele fosse razionalizzata e ne costituiscono quindi la
giustificazione teorica.
6.6.1 Abbassamento del punto di congelamento
Considerando un composto solido (soluto, composto 1) disciolto in un liquido (solvente,
composto 2) il diagramma di equilibrio liquido solido assume la forma gi discussa e
riportata in Figura 43. Lasse delle ordinate con x
1
= 0 corrisponde al solvente puro.
Partendo da uno stato in cui vi il solvente puro liquido (punto A), la sua temperatura di
congelamento (in cui ovviamente si separa il solvente puro solido) quella corrispondente
al punto B. Se nel solvente di partenza si scioglie una piccola quantit di soluto (punto C) e
si abbassa la temperatura, il solvente puro solido si separa quando si giunge al punto D,
cio a una temperatura pi bassa di quella di congelamento del solvente puro. Leffetto
netto dellaggiunta di una piccola quantit di soluto a un solvente quindi quello di
abbassare la temperatura a cui inizia a solidificare il solvente
154
.
Se la concentrazione del soluto piccola ( 0
1
x ) quella del solvente praticamente
unitaria ( 1
2
x ) e quindi anche il suo coefficiente di attivit ( 1
2
). La relazione di
equilibrio (702) per il solvente diventa quindi:
|
|
.
|
\
|
=
|
|
.
|
\
|
=
|
|
.
|
\
|
=
2
, 2
2
2 , 2
2
, 2
2
1 1
) ln(
f
f fus
f
f fus
f
fus
TT
T
R
h
TT
T T
R
h
T T R
h
x (706)
dove si indicato con T T T
f f
=
2
labbassamento della temperatura di congelamento
del solvente. Introducendo le approssimazioni
1 2
) ln( x x
155
e
2
2 2 f f
T TT
156
si ottiene:
154
Questo effetto ampiamente sfruttato nella pratica, per esempio per sciogliere la neve nelle strade.
155
Questa approssimazione corretta nel limite 1
2
x . Infatti, espandendo in serie di Taylor la
funzione ) ln(
2
x intorno al punto 1
2
= x si ottiene una espressione approssimata di ) ln(
2
x valida
solo nellintorno di 1
2
= x : ( )
1 2 2
1
2
2
1
2 2
1 1
) ln(
) ln( ) ln(
2
2
x x x
dx
x d
x x
x
x
= = +
=
=
.
156
Questa approssimazione lecita in quanto labbassamento della temperatura di congelamento, per
soluzioni diluite, solitamente piccolo.
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
261
|
|
.
|
\
|
2
2
, 2
1
f
f fus
T
T
R
h
x (707)
Figura 43 Diagramma di solubilit isobaro liquido solido per un sistema soluto (1)
solvente (2).
Dalla relazione precedente si ricava facilmente unespressione per labbassamento della
temperatura di congelamento del solvente:
fus
f
f
h
x RT
T
, 2
1
2
2
(708)
0 0.1 0.2 0.3 0.4 0.5 0.6 0.7 0.8 0.9 1
240
260
280
300
320
340
360
380
x
1
T
[
K
]
A
B
D
C
curva di congelamento
(precipita il solvente)
curva di solubilit
(precipita il soluto)
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
262
che stabilisce come labbassamento della temperatura di congelamento non dipenda dal tipo
di soluto e vari linearmente con la frazione molare del soluto. Questa nota come legge
dellabbassamento della temperatura di congelamento di vant Hoff
157
. I valori del rapporto
fus f
h RT
, 2
2
2
sono caratteristici di ciascun solvente e sono spesso tabulati sotto il nome di
costanti crioscopiche
158
.
6.6.2 Abbassamento della tensione di vapore
Considerando una miscela liquida di due composti di cui uno (soluto, composto 1) non sia
presente nella fase vapore in equilibrio, mentre laltro (solvente, composto 2) s
159
, la
relazione generale di equilibrio liquido vapore (653) valida per sistemi a bassa media
pressione pu essere scritta solo per il composto 2
160
:
( ) ( ) x , ,
2 2 2
P T x T P P
o
(709)
in cui si considerato che la frazione molare del composto 2 in fase vapore unitaria.
Se la concentrazione del soluto piccola ( 0
1
x ) quella del solvente praticamente
unitaria ( 1
2
x ) e quindi anche il suo coefficiente di attivit ( 1
2
). La relazione di
equilibrio per il solvente diventa quindi:
( ) ( )( )
1 2 2 2
1 x T P x T P P
o o
= (710)
Si nota da questa relazione che la pressione di equilibrio in presenza di un soluto, P, risulta
inferiore a quella del solvente puro, P
o
, in quanto x
2
<1. La presenza di un soluto quindi
diminuisce la tensione di vapore del solvente di un valore pari a:
( ) ( )
1 2 2
x T P P T P P
o o o
= (711)
che stabilisce come labbassamento della tensione di vapore non dipenda dal tipo di soluto
e vari linearmente con la frazione molare del soluto. Questa nota come legge
dellabbassamento della tensione di vapore di Raoult.
157
Jacobus Hendricus van't Hoff, 1852 - 1911, chimico fisico olandese.
158
Bisogna, come al solito, prestare attenzione alle unit di misura. Per esempio, spesso i valori
tabulati vanno utilizzati esprimendo la concentrazione del soluto non come frazione molare ma come
molalit.
159
Questa la situazione usuale di un solido (soluto) disciolto in un liquido (solvente).
160
In quanto solo il composto 2 presente in entrambe le fasi.
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
263
6.6.3 Innalzamento della temperatura di ebollizione
Considerando, analogamente al paragrafo precedente, una miscela liquida di due composti
di cui solo uno (solvente, composto 2) presente nella fase vapore in equilibrio, risulta
immediato dedurre che poich la presenza di un soluto abbassa la tensione di vapore del
solvente, esso ne innalza anche la temperatura di ebollizione.
La relazione generale di equilibrio liquido vapore pu essere scritta solo per il solvente e
pu essere posta nella forma:
) , ( ) , , ( ) , ( ) , , (
2 2 2 2 2
P T f P T x P T f P T f
V L L
= = x x (712)
Utilizzando un approccio analogo a quello discusso nel paragrafo 4.7.3 per il calcolo della
fugacit di un solido puro
161
si pu ricavare:
( ) ( )
( )
|
|
.
|
\
|
|
|
.
|
\
|
=
=
|
|
.
|
\
|
|
|
.
|
\
|
T T R
h
P T f
T T R
h
P T f P T f
b
ev
L
b
cond
L V
1 1
exp ,
1 1
exp , ,
2
, 2
2
2
, 2
2 2
(713)
che, introdotta nella relazione precedente, porta per sistemi diluiti (in cui al solito 0
1
x ,
1
2
x e quindi anche 1
2
) alla:
|
|
.
|
\
|
=
|
|
.
|
\
|
=
|
|
.
|
\
|
=
2
, 2
2
2
, 2
2
, 2
2
1 1
) ln(
b
b
ev
b
b
ev
b
ev
TT
T
R
h
TT
T T
R
h
T T R
h
x (714)
dove si indicato con
2 b b
T T T = linnalzamento della temperatura di ebollizione del
solvente.
Con passaggi analoghi a quelli discussi nel paragrafo 6.6.1 la relazione precedente pu
essere manipolata per giungere alla seguente equazione:
ev
b
b
h
x RT
T
, 2
1
2
2
(715)
che stabilisce come linnalzamento della temperatura di ebollizione non dipenda dal tipo di
soluto e vari linearmente con la frazione molare del soluto. Questa nota come legge
dellinnalzamento della temperatura di ebollizione di vant Hoff. I valori del rapporto
161
Si lascia per esercizio allo studente la derivazione di questa relazione e delle seguenti.
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
264
ev b
h RT
, 2
2
2
sono caratteristici di ciascun solvente e sono spesso tabulati sotto il nome di
costanti ebullioscopiche
162
.
6.6.4 Ripartizione di un soluto in due solventi
Se un terzo composto (soluto, indicato come composto 3) viene aggiunto a un sistema
costituito da due liquidi completamente immiscibili (solventi, costituiti dal composto 1 che
forma la fase e composto 2 che forma la fase , rispettivamente), esso si ripartisce tra le
due fasi
163
.
La relazione generale di equilibrio liquido liquido pu essere scritta quindi solo per il
soluto e pu essere posta nella forma:
) , , (
) , , (
3 3
x x P T f P T f
L L
= (716)
Utilizzando lapproccio indiretto per valutare la fugacit si ottiene:
) , , ( ) , ( ) , , ( ) , (
3 3 3 3 3 3
x x P T x P T f P T x P T f
L L
= (717)
Se la concentrazione del soluto piccola ( 0 ; 0
3 3
x x ) quella dei solventi nelle due
fasi praticamente unitaria ( 1 ; 1
2 1
x x ) e quindi i coefficienti di attivit del soluto
nelle due fasi sono uguali a quelli a diluizione infinita (
= =
,
3 3
,
3 3
;
). La
relazione di equilibrio per il soluto diventa quindi:
3
,
3
,
3
3
3
) , (
) , (
K
P T
P T
x
x
= =
(718)
che stabilisce come la ripartizione (cio il rapporto tra le concentrazioni) di un soluto in
due solventi sia costante a una data temperatura e pressione. Questo noto come legge di
ripartizione di Nernst
164
. I valori del rapporto
=
,
3
,
3 3
K sono caratteristici di
ciascun sistema soluto solvente solvente e sono spesso tabulati sotto il nome di
coefficienti di ripartizione.
162
Per i valori tabulati delle costanti ebullioscopiche valgono le stesse raccomandazioni fatte per
quelli delle costanti crioscopiche.
163
Si assume che il composto aggiunto sia solubile in entrambi i liquidi; si vengono cos a formare
due fasi: la fase costituita dai composti 1 e 3, e la fase costituita dai composti 2 e 3. Questo viene
utilizzato nella pratica industriale e di laboratorio dellestrazione liquido - liquido.
164
Walther Hermann Nernst, 1864 1941, fisico tedesco.
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
265
6.6.5 Solubilit dei gas nei liquidi
Considerando una miscela liquida di due composti di cui uno (il gas, composto 1)
supercritico, mentre laltro (il liquido, composto 2) subcritico, la relazione generale di
equilibrio liquido vapore (684) :
( ) ( )
( )
( ) P T
P T
x P T H P T Py
V
,
, ,
, , ,
1
1
1 1 1 1
=
x
y
(719)
Se la concentrazione del gas nel liquido piccola ( 0
1
x ) il suo coefficiente di attivit
uguale a quello a diluizione infinita (
=
1 1
). In questo caso, se si assume anche il gas
perfetto, la relazione di equilibrio per il soluto assume la forma nota come legge di Henry:
( )
1 1 1
, x P T H Py = (720)
6.7 Esercizi
Calcolare la pressione di bolla e la composizione della fase vapore in equilibrio per una
miscela liquida contenente il 25% di 2-propanolo (composto 1) in acqua (composto 2) a
353,15 [K]. I parametri del modello di Wilson per questo sistema sono:
7292 , 0 ; 1258 , 0
21 12
= = . I parametri dellequazione di Antoine ( ) C T B A P = ln ,
T in [K] e P in [kPa], per il 2-propanolo e lacqua sono rispettivamente A = 16,6780 e
16,2887; B = 3640,20 e 3816,44; C = 53,54 e 46,13.
Risultato: P = 91,5 [kPa]; y
1
= 0,538.
Calcolare la pressione di rugiada e la composizione della fase liquida in equilibrio per una
miscela vapore contenente il 60% di 2-propanolo (composto 1) in acqua (composto 2) a
353,15 [K]. I parametri del modello di Wilson per questo sistema sono:
7292 , 0 ; 1258 , 0
21 12
= = . I parametri dellequazione di Antoine ( ) C T B A P = ln ,
T in [K] e P in [kPa], per il 2-propanolo e lacqua sono rispettivamente A = 16,6780 e
16,2887; B = 3640,20 e 3816,44; C = 53,54 e 46,13.
Risultato: P = 96,7 [kPa]; x
1
= 0,449.
Calcolare la temperatura di bolla e la composizione della fase vapore in equilibrio per una
miscela liquida contenente l85% di 2-propanolo (composto 1) in acqua (composto 2) a
101,33 [kPa]. I parametri del modello di Wilson per questo sistema sono:
7292 , 0 ; 1258 , 0
21 12
= = . I parametri dellequazione di Antoine ( ) C T B A P = ln ,
T in [K] e P in [kPa], per il 2-propanolo e lacqua sono rispettivamente A = 16,6780 e
16,2887; B = 3640,20 e 3816,44; C = 53,54 e 46,13.
Risultato: T = 354,0 [K]; y
1
= 0,815.
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
266
Calcolare la temperatura di rugiada e la composizione della fase liquida in equilibrio per
una miscela vapore contenente il 40% di 2-propanolo (composto 1) in acqua (composto 2) a
101,33 [kPa]. I parametri del modello di Wilson per questo sistema sono:
7292 , 0 ; 1258 , 0
21 12
= = . I parametri dellequazione di Antoine ( ) C T B A P = ln ,
T in [K] e P in [kPa], per il 2-propanolo e lacqua sono rispettivamente A = 16,6780 e
16,2887; B = 3640,20 e 3816,44; C = 53,54 e 46,13.
Risultato: T = 360,6 [K]; x
1
= 0,064.
Calcolare la pressione di bolla e la composizione della fase vapore in equilibrio per una
miscela vapore contenente il 20% di propilene (composto 1) in n-butano (composto 2) a
350 [K] utilizzando lEoS PR.
Risultato: P = 13,54 [bar]; y
1
= 0,383.
Calcolare la pressione di bolla e la composizione della fase vapore in equilibrio con due fasi
liquide contenenti isobutano (composto 1) e furfurolo (composto 2) a 37,8 [C]. A questa
temperatura la tensione di vapore dei due composti vale 4,956 e 0,005 [bar] per lisobutano
e il furfurolo, rispettivamente. La frazione molare di isobutano nelle due fasi liquide vale
0,1128 e 0,9284. I parametri del modello di van Laar valgono 62 , 2
'
12
= A e 02 , 3
'
21
= A .
Risultato: P = 4,69 [bar]; y
1
= 0,999.
Calcolare la frazione molare di acetone (composto 1) in due solventi che possono essere
considerati in prima approssimazione tra loro immiscibili: diclorometano (composto 2) e
acqua (composto 3)
165
. I parametri del modello di van Laar valgono 37 , 0
'
12
= A ;
34 , 0
'
21
= A ; 29 , 2
'
13
= A ; 35 , 1
'
31
= A .
Risultato: 444 , 0 ; 063 , 0
1 1
= =
x x .
Calcolare la temperatura di eutettico del sistema p-xilene m-xilene a pressione ambiente.
Lentalpia di fusione e la temperatura di fusione normale dei due composti valgono:
2764,93 e 4089,92 [cal/mol]; - 47,9 e 13,9 [C] per m-xilene e p-xilene, rispettivamente.
Risultato: T
E
= 220,8 [K].
165
In realt, in queste condizioni la solubilit del diclorometano in acqua, ma soprattutto dellacqua
nel diclorometano non trascurabile. I valori calcolati saranno quindi validi solo in prima
approssimazione.
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
267
Calcolare la frazione molare dellossigeno disciolto in acqua a contatto con latmosfera. La
costante di Henry dellossigeno in acqua a temperatura ambiente vale 4,4 10
4
[bar].
Risultato: x = 4,8 10
-6
.
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
269
7 Sistemi reagenti
Fino a questo momento si sono considerati sistemi non reagenti, in cui cio gli atomi che
costituiscono una data molecola rimangono sempre in quella stessa molecola. Questo non
ovviamente il caso pi generale possibile, in particolare per un ingegnere chimico che si
interessa per definizione della trasformazione della materia
166
che comporta una
riorganizzazione degli atomi allinterno di molecole diverse
167
.
Lobiettivo di questo capitolo quello di dedurre delle relazioni che consentano di calcolare
la composizione di un sistema in condizioni di equilibrio sotto il vincolo di conservazione
della materia a livello di specie atomiche. In altri termini, dato un certo numero di moli di
alcune specie che si assumono presenti nel sistema
168
si vuole calcolare il numero di moli di
tutte queste specie nelle condizioni di equilibrio, cio come si ridistribuiscono gli atomi tra
le specie presenti. Questo comporta la necessit di riformulare le equazioni di bilancio per
sistemi multicomponente, in quanto il termine di produzione presente nei bilanci di materia
pu essere diverso da zero.
Al termine dello studio di questo capitolo lo studente dovrebbe conoscere:
i bilanci di materia per sistemi multicomponente reagenti e non;
il vincolo stechiometrico;
il grado di avanzamento per sistemi chiusi semplici;
la relazione di equilibrio per sistemi chiusi semplici;
la definizione di
o
R
G e
o
R
H ;
la definizione di costante di equilibrio;
lo stato di riferimento utilizzato per il calcolo dellentalpia e dellenergia libera di
Gibbs in sistemi reagenti;
166
La trasformazione della materia avviene attraverso delle trasformazioni chimiche ed loggetto di
studio della chimica. Lingegnere chimico interessato alle trasformazioni chimiche per quantificarne
gli effetti ai fini della progettazione della apparecchiature industriali in cui esse vengono condotte: i
reattori chimici.
167
Per esempio, nel caso di metano e ossigeno che si trasformano in anidride carbonica e acqua
latomo di carbonio inizialmente presente nella molecola di metano si ritrova poi in quella di anidride
carbonica.
168
Si noti che lassunzione di quali specie sono presenti nel sistema condiziona i risultati dei calcoli.
Per esempio, si troveranno risultati completamente differenti se si considerano presenti in un sistema
metano e ossigeno oppure metano, ossigeno, acqua e anidride carbonica. Nel primo caso non si pu
avere alcuna trasformazione della materia (cio alcuno scambio di atomi tra molecole diverse) in
quanto gli atomi di carbonio e idrogeno possono stare solo nella molecola di metano e quelli di
ossigeno solo nella molecola di ossigeno. Nel caso secondo invece si pu avere trasformazione di
metano e ossigeno in anidride carbonica e acqua (o viceversa).
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
270
gli stati di riferimento standard utilizzati per il calcolo dellattivit e del
o
R
G per
composti in fase gas, liquida e solida;
linfluenza della temperatura sulla costante di equilibrio;
le relazioni di equilibrio per sistemi chiusi complessi;
le relazioni di equilibrio per sistemi aperti semplici e complessi;
il bilancio di energia per sistemi multicomponente reagenti e non.
7.1 Bilancio di materia in sistemi multicomponente
Come discusso in precedenza, il bilancio di materia la traduzione in termini matematici
dellaffermazione che la somma dei flussi netti entranti in un sistema e della quantit netta
prodotta nellunit di tempo allinterno del sistema stesso deve essere uguale alla variazione
netta nel tempo della quantit presente allinterno del sistema stesso, come formalizzato
nella relazione generale (25).
Nel caso di miscele possibile applicare questa relazione generale al numero di moli di
ciascuna specie presente nel sistema:
i OUT i IN i
i
G n n
dt
dn
&
& & + =
, ,
(721)
dove
i
G
&
[mol s
-1
] rappresenta la velocit di produzione molare del composto i-esimo
allinterno del sistema dovuto esclusivamente alle trasformazioni chimiche. Moltiplicando
per il peso molecolare (costante) del composto i-esimo il bilancio precedente pu essere
riscritto in termini massici come:
i OUT i IN i
i
M m m
dt
dm
&
& & + =
, ,
(722)
dove
i
M
&
[kg s
-1
] rappresenta la velocit di produzione massica del composto i-esimo
allinterno del sistema dovuto esclusivamente alle trasformazioni chimiche.
Sommando le relazioni precedenti su tutti i composti presenti nel sistema si ottengono i
bilanci di materia totali:
=
+ =
N
i
i OUT IN
G n n
dt
dn
1
&
& & (723)
OUT IN
m m
dt
dm
& & = (724)
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
271
Si noti che il termine di produzione massica totale sempre nullo (almeno per le situazioni
normalmente contemplate nellambito dellingegneria chimica
169
), mentre non lo quello di
produzione molare totale in quanto il riarrangiamento degli atomi in molecole diverse pu
far aumentare o diminuire il numero di moli totali
170
.
In realt, nellambito della termodinamica non possibile utilizzare i bilanci di materia
nella forma completa presentata sopra in quanto la termodinamica non si occupa della
velocit con cui un sistema evolve ma solo dello stato finale di equilibrio a cui giunge. I
bilanci completi rappresentati dalle equazioni (721) - (724) viceversa descrivono la
dinamica di un sistema e richiedono la conoscenza della velocit di produzione del
composto i-esimo allinterno del sistema dovuto esclusivamente alle trasformazioni
chimiche, cio della velocit delle reazioni chimiche. Il calcolo di tale velocit esula
dallambito di applicazione della termodinamica ed affrontato nellambito della cinetica
chimica.
Nellambito della termodinamica si possono per utilizzare due forme semplificate dei
bilanci di materia: quello per sistemi chiusi e quello in condizioni stazionarie.
Per sistemi chiusi i bilanci precedenti divengono:
i
i
G
dt
dn
&
= (725)
i
i
M
dt
dm
&
= (726)
=
=
N
i
i
G
dt
dn
1
&
(727)
0 =
dt
dm
(728)
che possono essere posti nella forma integrata tra un dato stato iniziale e lo stato di
equilibrio:
i
o
i i i
n
n
i i i
G n n dt G dn dt G dn
i
o
i
= = =
0
& &
(729)
169
In altri termini, gli atomi si possono riarrangiare in molecole diverse, ma non si possono creare o
distruggere.
170
Per esempio, carbonio e ossigeno che si trasformano in anidride carbonica comportano un
riduzione del numero di moli totali, anche se la massa totale [kg] non varia.
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
272
i
o
i i i
m
m
i i i
M m m dt M dm dt M dm
i
o
i
= = =
0
& &
(730)
= =
= = =
N
i
i
o
N
i
i
n
n
N
i
i
G n n dt G dn dt G dn
o
1
0
1 1
& &
(731)
0
0 0
0
m m dm dm
i
m
m
= = =
(732)
dove G
i
[mol] rappresenta il numero di moli del composto i-esimo che devono essere
prodotte allinterno del sistema attraverso le trasformazioni chimiche per raggiungere le
condizioni di equilibrio. Analogamente, M
i
[kg] rappresenta la massa del composto i-esimo
che deve essere prodotta. In queste relazioni non pi presente la velocit di produzione,
ma solo lo stato iniziale e finale e quindi la loro applicazione rientra nellambito della
termodinamica.
Per sistemi in condizioni stazionarie le relazioni di bilancio divengono:
0
, ,
= +
i OUT i IN i
G n n
&
& & (733)
0
, ,
= +
i OUT i IN i
M m m
&
& & (734)
0
1
= +
=
N
i
i OUT IN
G n n
&
& & (735)
0 =
OUT IN
m m & & (736)
Queste relazioni trovano applicazione nellambito della termodinamica se si assume che
nelle condizioni di uscita sia raggiunto lequilibrio termodinamico. In questo caso la
velocit di produzione che compare nelle equazioni di bilancio pu essere interpretata come
la variazione delle portate tra ingresso e uscita dal sistema necessaria a portare un sistema
dalle condizioni di ingresso alle condizioni di equilibrio. Anche in queste relazioni non
pi quindi presente la reale velocit di produzione, ma solo due stati, rappresentati dalle
condizioni in ingresso e da quelle di equilibrio (in uscita). La applicazione di queste
relazioni rientra quindi ancora nellambito della termodinamica.
7.1.1 Sistemi non reagenti
Per sistemi multicomponente non reagenti
171
le relazioni precedenti si semplificano in
quanto il termine di generazione risulta nullo. In questo caso non mai necessario
171
Sistemi non reagenti sono quelli in cui non avvengono trasformazioni chimiche.
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
273
conoscere la velocit delle trasformazioni chimiche e quindi le relazioni generali seguenti si
possono sempre applicare nellambito della termodinamica:
OUT i IN i
i
n n
dt
dn
, ,
& & = (737)
OUT i IN i
i
m m
dt
dm
, ,
& & = (738)
OUT IN
n n
dt
dn
& & = (739)
OUT IN
m m
dt
dm
& & = (740)
In condizioni stazionarie le equazioni di bilancio diventano:
OUT i IN i
n n
, ,
& & =
(741)
OUT i IN i
m m
, ,
& & =
(742)
OUT IN
n n & & = (743)
OUT IN
m m & & =
(744)
mentre per sistemi chiusi:
0 = = = =
dt
dm
dt
dn
dt
dm
dt
dn
i i
(745)
7.2 Sistemi chiusi
Come detto in precedenza i due stati considerati possono essere quello iniziale e quello
finale (di equilibrio) in un sistema chiuso o quello di ingresso e quello di uscita (di
equilibrio) per un sistema aperto in condizioni stazionarie. Nel seguito si discuter il caso di
sistemi chiusi. Le conclusioni a cui si giunger valgono, in termini generali, anche per i
sistemi aperti in condizioni stazionarie, come verr discusso pi avanti.
7.2.1 Il vincolo stechiometrico
La conservazione della materia a livello delle singole specie atomiche implica che alcuni
vincoli devono essere soddisfatti. Questi vincoli nascono dallaffermazione che il numero
di moli di una specie atomica presenti nei due stati che si considerano devono essere gli
stessi. Quindi, se inizialmente sono presenti nel sistema per esempio 10 atomi di carbonio,
nello stato di equilibrio si dovranno ritrovare gli stessi 10 atomi di carbonio, eventualmente
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
274
distribuiti in molecole diverse da quelle in cui erano presenti inizialmente. Questa
affermazione pu essere formalizzata nelle seguenti relazioni, una per ciascuna specie
atomica presente il cui numero di moli deve rimanere costante:
at k
N
i
o
i ki
N
i
i ki
N k b n a n a ... 1
1 1
= = =
= =
(746)
dove a
ki
il numero di atomi di tipo k presenti nella molecola della specie i, lapice
o
indica
le condizioni iniziali e b
k
il numero di moli di atomi k presenti inizialmente. N
at
il
numero di atomi diversi presenti nel sistema. Queste relazioni possono essere scritte nella
forma vettoriale compatta:
b An An
o
= =
(747)
dove A la matrice atomi - specie con elementi a
ki
, n il vettore del numero di moli delle
specie presenti e b il vettore del numero di moli iniziale delle specie atomiche. Il vincolo
stechiometrico, rappresentato dalla relazione precedente, deriva dal fatto che b fissato e
costante. Tale vincolo impone che, dato un certo numero di moli iniziale delle specie
atomiche b (o, in altri termini, un dato numero di moli iniziali delle specie presenti, n
o
), il
numero di moli finale delle specie presenti non pu assumere un valore arbitrario ma deve
rispettare le N
at
relazioni (747).
Considerando a titolo di esempio un sistema chiuso in cui si assumono presenti CO, CO
2
e
O
2
e contenente inizialmente una mole di CO e due moli di O
2
, il vettore delle specie
presenti :
| |
T
O CO CO
2 2
= S (748)
dove lapice
T
indica il vettore trasposto. Questo vettore serve ad ordinare le specie da 1 a
N: in questo caso la specie 1 il CO, la specie 2 la CO
2
e la specie 3 lO
2
. Il vettore degli
atomi (che svolge una funzione analoga) invece:
| | C O = E (749)
Ne consegue che la matrice atomi specie in questo caso :
(
=
0 1 1
2 2 1
A (750)
dove le righe sono relative agli atomi (O e C, dallalto in basso) e le colonne alle specie
(CO, CO
2
e O
2
da sinistra a destra).
Il vettore del numero di moli, sulla base dellordinamento deciso per le specie, quindi:
| |
T
O CO CO
n n n
2 2
= n (751)
e quindi il vettore del numero di moli iniziali in questo caso :
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
275
| |
T
2 0 1 =
o
n (752)
Il numero di moli iniziale delle specie atomiche presenti in questo caso quindi pari a :
(
=
(
(
(
= =
1
5
2
0
1
0 1 1
2 2 1
o
An b (753)
Ne consegue che il vincolo stechiometrico assume la forma:
= +
= + +
=
(
(
(
1
5 2 2
1
5
0 1 1
2 2 1
2
2 2
2
2
CO CO
O CO CO
O
CO
CO
n n
n n n
n
n
n
(754)
che definisce la conservazione del numero di moli di ossigeno e di carbonio,
rispettivamente. Appare evidente che il numero di moli delle tre specie presenti nello stato
di equilibrio non pu assumere un valore qualsiasi ma deve rispettare le due equazioni
(754). In altri termini, il numero di variabili indipendenti del problema non pi pari al
numero di specie presenti (3 in questo esempio), ma al numero di specie presenti meno il
numero di vincoli rappresentati dalle equazioni (747). Nellesempio considerato il numero
di vincoli pari a 2 e quindi il numero di variabili indipendenti pari a 3 2 = 1. Ci
significa che una volta noto il valore del numero di moli di una specie, quello delle altre
due fissato dal vincolo stechiometrico e pu essere calcolato con le due relazioni (754).
Unimportante conseguenza dellequazione (747) che il vincolo stechiometrico non
dipende in modo biunivoco dal numero di moli iniziali delle specie presenti, ma bens dal
numero di moli iniziali degli atomi presenti. Questo significa che sistemi contenenti
inizialmente un diverso numero di moli delle specie molecolari ma lo stesso numero di moli
delle specie atomiche originano lo stesso vincolo stechiometrico e quindi raggiungono lo
stesso stato di equilibrio
172
.
Il numero di equazioni indipendenti che nascono dalle relazioni (747) solitamente pari al
numero di specie atomiche presenti nel sistema, N
at
. In realt, pi correttamente tale
numero, indicato nel seguito con NA, pari al rango della matrice elementi specie A
173
:
172
In altri termini, due sistemi chiusi contenenti inizialmente 1 mole di CO, 1 di CO
2
e 0 di O
2
,
oppure 2 moli di CO, 0 di CO
2
e 0,5 di O
2
, contengono entrambi 2 moli di atomi di C e 3 moli di
atomi di O e quindi conducono allo stesso vettore b. Lo stato di equilibrio che raggiungeranno i due
sistemi sar quindi lo stesso.
173
Per esempio, si verifica facilmente che un sistema contenente NH
3
, CO
2
, H
2
O e CO(NH)
2
coinvolge 4 specie atomiche (N, H, C e O) ma dai bilanci materiali su tali specie atomiche si
originano solo 3 equazioni indipendenti, in quanto in questo caso il rango della matrice elementi
specie pari appunto a 3.
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
276
) ( rank A = NA (755)
Come detto in precedenza, il numero di variabili indipendenti del problema, indicato nel
seguito con NR, pari al numero di specie presenti, N, meno il numero di equazioni
indipendenti che nascono dalle relazioni (747), NA (che solitamente, ma non sempre, pari
al numero di specie atomiche presenti):
NA N NR = (756)
Il calcolo del numero di moli di tutte le specie presenti in condizioni di equilibrio richiede
quindi la scrittura di NR equazioni per la valutazione del numero di moli di NR specie; il
numero di moli delle rimanenti NA specie possono poi essere calcolate dalle NA equazioni
indipendenti derivanti dal vincolo stechiometrico.
7.2.2 Il grado di avanzamento per sistemi semplici
Un metodo semplice per ridurre il numero delle variabili da N a NR tenendo implicitamente
conto delle NA equazioni di bilancio materiale delle specie atomiche quello cosiddetto del
grado di avanzamento. Per semplicit di esposizione viene considerato nel seguito il caso
particolare di NR = 1 (sistemi semplici); la generalizzazione al caso di NR > 1 (sistemi
complessi) verr discussa pi avanti.
La scrittura di una reazione chimica nella forma classica utilizzata nellambito della
chimica fornisce informazioni di tipo qualitativo. Per esempio la reazione:
2 2
CO O CO = + (757)
dice che monossido di carbonio e ossigeno possono trasformarsi in anidride carbonica (e
viceversa). Non fornisce invece alcuna informazione quantitativa. Questo avviene nel
momento in cui la reazione chimica viene bilanciata introducendo i coefficienti
stechiometrici:
2 2
2
1
CO O CO = + (758)
Un reazione chimica bilanciata impone che affinch i bilanci materiali sulle specie
atomiche siano rispettati (bilanciare una reazione chimica significa infatti imporre che il
numero di atomi di una data specie presenti tra i reagenti sia uguale al numero di atomi di
quella specie presenti tra i prodotti; questo non significa altro che imporre il vincolo
stechiometrico (747)) la variazione del numero di moli di ciascuna specie non pu essere
arbitrario ma deve rispettare certi vincoli (quelli stechiometrici, appunto)
174
. Lesempio
della reazione (758) indica che se scompare una mole di CO deve scomparire mezza mole
174
Imporre dei vincoli sulla variazione del numero di moli ovviamente lo stesso che imporre dei
vicoli sul numero di moli finali a fronte di un dato numero di moli iniziali.
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
277
di O
2
e deve formarsi una mole di CO
2
. In termodinamica quindi una reazione chimica pu
essere vista come una equazione stechiometrica che traduce il principio di conservazione
della materia delle specie atomiche. In questo senso risulta pi utile, anche se meno
intuitivo, scrivere una generica reazione chimica:
... ... + + = + + N M B A
N m B A
(759)
nella forma:
... ... 0 + + + + = N M B A
N m B A
(760)
cio:
=
=
N
j
j j
S
1
0 (761)
dove S
j
indicano le diverse specie e
j
i relativi coefficienti stechiometrici assunti con una
convenzione di segno: positivi per i prodotti e negativi per i reagenti
175
. In questo modo a
ogni equazione stechiometrica risulta associato un vettore stechiometrico i cui elementi
sono appunto i coefficienti stechiometrici. Per esempio, la reazione (758) viene scritta
come:
2 2
2
1
0 CO O CO + = (762)
a cui associato il vettore delle specie (che al solito serve solo per ordinarle):
| |
T
CO O CO
2 2
= S (763)
e il vettore stechiometrico:
T
(
+ = 1
2
1
1 (764)
Il vettore stechiometrico risulta utile per tradurre in una forma matematica semplice
laffermazione che la variazione del numero di moli delle specie presenti in una reazione
bilanciata non pu essere arbitrario, ma deve rispettare il vincolo stechiometrico.
Considerando per esempio la reazione (758), il vincolo indotto dal bilanciamento della
reazione impone che se scompare una mole di CO deve scomparire mezza mole di O
2
e
deve formarsi una mole di CO
2
. Questo pu essere espresso in termini matematici dalla
relazione:
175
Il coefficiente stechiometrico di un composto inerte, che cio non partecipa alla trasformazione
chimica, ovviamente uguale a zero.
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
278
2 2
2
CO O CO
n n n = =
(765)
avendo indicato con una generica variazione. Questa relazione coinvolge il vettore
stechiometrico in quanto pu essere messa nella forma:
2
2
2
2
CO
CO
O
O
CO
CO
n n
n
=
(766)
Una equazione stechiometrica impone quindi che, affinch siano rispettati i bilanci
materiali delle specie atomiche, il rapporto tra la variazione del numero di moli di una
specie e il relativo coefficiente stechiometrico sia uguale per tutte le specie presenti nella
reazione bilanciata. In termini differenziali questo conduce ala relazione:
N j d
dn
j
j
... 1 = =
(767)
che definisce il grado di avanzamento della reazione, . Considerando la variazione del
numero di moli tra lo stato di equilibrio e quello iniziale si ottiene la relazione generale:
j
o
j j j
n
n
j
n n d dn
j
o
j
= =
0
(768)
che pu essere espressa nella forma vettoriale compatta:
+ =
o
n n
(769)
Confrontando questa equazione con lespressione del bilancio materiale (729) appare
evidente che le due equazioni coincidono pur di definire il termine di generazione per
sistemi semplici come:
i i
G = (770)
Le equazioni (769) non rappresentano quindi altro che una forma dei bilanci materiali per le
singole specie che includono implicitamente i bilanci materiali per le specie atomiche.
Queste equazioni consentono di calcolare il numero di moli di tutte le specie presenti nella
razione bilanciata sulla base della conoscenza del numero di moli iniziali, del vettore
stechiometrico e della sola variabile . In altri termini, poich si sta discutendo il caso di
sistemi semplici (cio caratterizzati da NR=1), sufficiente conoscere il valore di una
variabile (, che coincide con la variazione del numero di moli di una specie con
coefficiente stechiometrico uguale a +1) per calcolare la variazione del numero di moli di
tutte le specie presenti. Il valore del grado di avanzamento pu essere calcolato da una
relazione dedotta dallimporre che il sistema raggiunga le condizioni di equilibrio.
Risulta infine importante sottolineare che lequazione stechiometrica che si utilizza deve
contenere tutte le specie presenti (a parte eventuali inerti), ma non rappresenta la reazione
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
279
che effettivamente avviene a livello molecolare. In termodinamica, come gi detto, essa
rappresenta semplicemente unequazione che traduce il vincolo della conservazione della
materia a livello di atomi. Lo studio delle reazioni chimiche che effettivamente avvengono
a livello molecolare lambito di studio della cinetica chimica.
7.2.3 Condizione di equilibrio per sistemi semplici
Le condizioni di equilibrio per un sistema soggetto al vincolo di mantenere costante la
temperatura e la pressione che lenergia libera di Gibbs sia minima
176
, il che implica che il
suo differenziale sia nullo:
( ) 0 min
, ,
=
P T P T
dG G
n
(771)
Inserendo la relazione (237) a T e P costanti si ottiene:
0
1
,
= =
=
N
i
i i P T
dn dG (772)
che, inserendo la relazione (767), diventa:
0
1 1 1
=
|
|
.
|
\
|
= =
= = =
N
i
i i
N
i
i i
N
i
i i
d d dn (773)
Poich la variazione infinitesima del grado di avanzamento arbitraria, la condizione di
equilibrio si riduce alla relazione:
0
1
=
=
N
i
i i
(774)
La definizione di fugacit (561) pu essere integrata tra lo stato del sistema e uno stato di
riferimento (che deve essere alla stessa temperatura del sistema e viene solitamente, anche
se non necessariamente, scelto come composto puro; lo stato di aggregazione e la pressione
vengono viceversa scelti in modo da rendere pi semplici i calcoli, come discusso pi
avanti):
176
Si noti che una volta che un sistema ha raggiunto le condizioni di equilibrio con una
trasformazione qualsiasi (e quindi non necessariamente a T e P costante), la sua energia libera di
Gibbs deve essere comunque minima rispetto a qualsiasi variazione del sistema (in particolare della
sua composizione, e quindi del numero di moli delle specie presenti) a quella T e P. Le conclusioni
che si trarranno partendo dal caso particolare di T e P costante sono quindi, al solito, del tutto
generali.
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
280
= =
=
x x , ,
1 , ,
, ,
1 , ,
ln
P T
x P T
i
P T
x P T
i
i r i r
f RTd d (775)
da cui si ottiene:
( ) ( )
( )
( )
( ) ( ) x
x
x , , , ln
,
, ,
ln , , ,
r i
r i
i
r i i
P P T a RT
P T f
P T f
RT P T P T =
|
|
.
|
\
|
= (776)
dove si introdotta la attivit, a
i
, definita come il rapporto tra la fugacit di un composto
nelle condizioni del sistema e la fugacit dello stesso composto nello stato di riferimento
scelto.
Inserendo questa relazione nella (774) si ottiene:
( ) ( ) ( ) ( ) 0 , , , ln , , ,
1 1 1
= + =
= = =
N
i
r i i
N
i
r i i
N
i
i i
P P T a RT P T P T x x (777)
Lultimo termine pu essere rimaneggiato algebricamente come segue
177
:
( ) ( ) ( ) ( )
( ) ( ) ( ) ( )
= =
= =
= =
= =
N
i
r i
N
i
r i
N
i
r i i
N
i
r i i
i i
P P T a RT P P T a RT
P P T a RT P P T a RT
1 1
1 1
, , , ln , , , ln
, , , ln , , , ln
x x
x x
(778)
che inserito nella relazione di equilibrio conduce alla equazione:
( ) ( ) ( ) 0 , , , ln ,
1 1
= +
= =
N
i
r i
N
i
r i i
i
P P T a RT P T
x (779)
Si noti che i due termini presenti in questa relazione non sono indipendenti in quanto
ciascun termine della sommatoria e della produttoria ha in comune lo stato di riferimento,
che deve ovviamente essere lo stesso per i termini omologhi. quindi possibile scegliere
uno stato di riferimento diverso per le diverse specie presenti, purch lo si mantenga uguale
nel calcolo del potenziale chimico e dellattivit della stessa specie.
Il primo termine della relazione (779) pu essere calcolato ricordando che il potenziale
chimico di un composto puro coincide con lenergia libera di Gibbs molare. Di
177
Il simbolo
N
N
i
i
x x x x =
=
...
2 1
1
indica, analogamente alla sommatoria, la produttoria degli N
termini.
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
281
conseguenza la sommatoria viene solitamente indicata come la variazione dellenergia
libera della reazione,
o
R
G :
( ) ( ) ( )
= =
= =
N
i
r i i
N
i
r i i r
o
R
P T g P T P T G
1 1
, , , (780)
Il significato fisico del
o
R
G evidente dalla relazione precedente: esso indica la variazione
di energia libera di Gibbs associata alla trasformazione completa dei reagenti presenti in
quantit stechiometriche nei prodotti nelle condizioni di riferimento. Per esempio, il
o
R
G
della reazione (758) pari a
2 2
2 1
CO O CO
o
R
g g g G + = , cio alla variazione di energia
libera di Gibbs che si ha quando una mole di CO e mezza mole di O
2
si trasformano in una
mole di CO
2
.
Il calcolo dellenergia libera di Gibbs molare delle diverse specie richiede la definizione di
uno stato di riferimento comune per tutte le specie chimiche, in quanto uno stesso atomo
pu far parte di diverse specie, come reagente prima e come prodotto poi. Il riferimento
solitamente scelto per il calcolo dellenergia libera di Gibbs molare quello degli elementi
chimici nella forma pi stabile nello stato di riferimento (cio alla temperatura del sistema e
alla pressione di riferimento), cos che il
o
R
G della reazione di formazione di un composto
dagli elementi, scritta assumendo che si formi una mole del composto stesso, coincide con
lenergia libera di Gibbs molare del composto stesso. Considerando per esempio la reazione
di formazione del CO dagli elementi:
CO O C = +
2
2
1
(781)
il
o
R
G di questa reazione viene calcolato, ricordando che il riferimento scelto implica che
lenergia libera di Gibbs degli elementi nello stato di riferimento sia nulla, come:
CO CO O C
o
CO f
o
R
g g g g g G = + = =
2
2 1
,
(782)
dove con
o
i f
g
,
si indicata lenergia libera di Gibbs di formazione del composto i. La
relazione operativa per il calcolo del
o
R
G diviene quindi la seguente:
( ) ( )
=
=
N
i
r
o
i f i r
o
R
P T g P T G
1
,
, , (783)
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
282
I valori dellenergia libera di Gibbs di formazione di molti composti sono tabulati
178
in uno
stato di riferimento standard a 298,15 [K] e 1 [bar] ed alcuni di essi sono riportati nella
Tabella 18.
Comunque, una volta fissato lo stato di riferimento il
o
R
G un numero che pu essere
facilmente calcolato in quanto non dipende n dalla pressione n dalla composizione del
sistema, ma solo dalla temperatura del sistema.
composto stato ( ) [bar] 1 , [K] 15 , 298
o
f
h
( ) [bar] 1 , [K] 15 , 298
o
f
g
acqua G -241818 -228572
acqua L -285830 -237129
ammoniaca G -45940 -16401
anidride carbonica G -393509 -394359
anidride solforica G -395720 -371060
anidride solforosa G -296830 -300194
carbonato di calcio S -1206920 -1128790
etano G -83820 -31855
metano G -74520 -50460
monossido di carbonio G -110525 -137169
ossido di calcio S -635090 -604030
Tabella 18 Entalpie ed energie libere di Gibbs di formazione molare (riferite cio a 1
mole di composto formato) standard in [J mol
-1
]. Stato standard per i composti gassosi:
gas perfetto puro a 298,15 [K] e 1 [bar]; per i composti liquidi e solidi: composto puro
liquido o solido a 298,15 [K] e 1 [bar].
Il secondo termine della relazione (779) pu essere calcolato utilizzando le relazioni
discusse nei capitoli precedenti per il calcolo della fugacit dei composti puri e in miscela.
Solitamente la produttoria viene indicata come la costante di equilibrio della reazione, K:
( ) ( )
=
=
N
i
r i
i
P P T a K
1
, , ,
x (784)
La costante di equilibrio dipende dalla composizione del sistema e quindi, attraverso le
relazioni (769), dal grado di avanzamento della reazione. Fissate temperatura, pressione e
stato di riferimento la relazione:
178
Si veda per esempio The NBS tables of chemical thermoodynamic properties, J. of Physical and
Chemical Reference Data, vol. 11, supp. 2 (1982).
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
283
( ) ( )
( )
|
|
.
|
\
|
= =
=
RT
P T G
P P T a K
r
o
R
N
i
r i
i
,
exp , , ,
1
(785)
rappresenta unequazione nella sola incognita che pu essere risolta
179
. Noto , attraverso
le relazioni (769) possibile calcolare il numero di moli di tutte le specie e quindi la
composizione in termini di frazione molare
180
.
7.2.4 Stati di riferimento standard
Come discusso in precedenza la scelta dello stato di riferimento completamente arbitraria
e pu essere diversa per ciascun composto, purch si utilizzi lo stesso stato di riferimento
per un dato composto per il calcolo sia dellattivit sia del
o
R
G . Daltro canto la scelta di
un opportuno stato di riferimento pu rendere pi agevoli i calcoli o il reperimento delle
informazioni termodinamiche. Di seguito sono riassunti alcuni degli stati di riferimento
standard pi utilizzati. Per ciascun caso si riportano anche le espressioni della costante di
equilibrio nellipotesi che tutti i composti siano nella stessa fase; nel caso in cui dei
composti siano presenti in fasi diverse si utilizzeranno le relative espressioni dellattivit.
7.2.4.1 Composti in fase gas nello stato del sistema
In questo caso lo stato di riferimento pi utilizzato gas perfetto a P
r
= 1 [bar]. La fugacit
nello stato di riferimento quindi uguale a 1 [bar] e lattivit si calcola come:
( ) ( )
[bar] 1
, ,
[bar] 1
, ,
i
G
i
G
i
i
Px P T P T f
a
x x
= = (786)
Il termine unitario al denominatore pu essere omesso pur di ricordare che la pressione al
numeratore deve essere espressa in [bar]:
179
Si tratta solitamente di unequazione algebrica non lineare che deve essere risolta numericamente e
che pu presentare pi di una radice, di cui solitamente solo una ha un significato fisicamente
accettabile. Il fatto che una radice dellequazione abbia senso fisico o meno non pu essere dedotto
immediatamente (il grado di avanzamento pu assumere a priori qualsiasi valore, positivo o negativo)
ma discende dal fatto che le grandezze fisiche calcolate con un dato valore del grado di avanzamento
siano accettabili o meno. Per esempio, il numero di moli non pu essere negativo e le frazioni molari
devono essere comprese tra 0 e 1.
180
Per quanto sia banale, assai importante ricordare che nel caso in cui siano presenti pi fasi la
frazione molare di un composto in una fase data dal rapporto tra il numero di moli del composto
presenti nella fase considerata e il numero di moli totale presenti nella stessa fase. Analogamente, se
in una fase presente un composto inerte il suo numero di moli deve ovviamente essere computato
nel numero di moli totali presenti in quella fase.
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
284
( )
i
G
i i
Px P T a x , ,
= (787)
Se la miscela gassosa ideale la relazione si semplifica nella:
( )
i
G
i i
Px P T a , = (788)
Se poi il gas assimilabile a un gas perfetto la relazione precedente diventa:
i i
Px a = (789)
e la costante di equilibrio assume la nota forma di produttoria delle pressioni parziali:
( )
=
=
N
i
i
i
Px K
1
(790)
Le frazioni molari possono essere espresse in funzione del grado di avanzamento tramite le
relazioni (769):
=
+
+
= =
N
j
j
o
j
i
o
i
T
i
i
n
n
n
n
x
1
(791)
e quindi la relazione di equilibrio (785) diventa:
( )
|
|
.
|
\
|
=
|
|
|
.
|
\
|
+
+
=
|
|
|
.
|
\
|
+
+
=
=
=
=
RT
T G
n
n
P
n
n
P
o
R
N
i
N
j
j
o
j
i
o
i
N
i
N
j
j
o
j
i
o
i
i i
1 ,
exp
1
1
1
1
(792)
dove si indicato con
=
=
N
i
i
1
la somma dei coefficienti stechiometrici. Si nota
immediatamente che la pressione del sistema influenza le condizioni di equilibrio solo nel
caso in cui 0
1
=
=
N
i
i
. Noto il
o
R
G (oltre ovviamente alla temperatura e pressione
del sistema e ai vettori del numero di moli iniziali e stechiometrico) questa equazione pu
essere risolta nellunica incognita .
7.2.4.2 Composti in fase liquida nello stato del sistema
In questo caso lo stato di riferimento pi utilizzato composto liquido puro a P
r
= 1 [bar].
Lattivit si calcola quindi come:
( )
( )
( ) ( )
( ) ] r [ba 1 ,
, , ,
] r [ba 1 ,
, ,
=
=
=
=
P T f
P T x P T f
P T f
P T f
a
L
i
i i
L
i
L
i
L
i
i
x x
(793)
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
285
Nel caso in cui la correzione di Poynting sia trascurabile la relazione si semplifica nella:
( ) x , , P T x a
i i i
= (794)
Se poi la miscela ideale si ottiene:
i i
x a = (795)
e la costante di equilibrio assume la nota forma di produttoria delle frazioni molari:
( )
=
=
N
i
i
i
x K
1
(796)
Introducendo la relazione (791) la relazione di equilibrio (785) diventa:
( )
|
|
.
|
\
|
=
|
|
|
.
|
\
|
+
+
=
=
RT
T G
n
n
o
R
N
i
N
j
j
o
j
i
o
i
i
1 ,
exp
1
1
(797)
da cui si nota, coerentemente con lipotesi di trascurare la correzione di Poynting, che la
pressione del sistema non influenza le condizioni di equilibrio. Anche in questo caso, noto
il
o
R
G (oltre ovviamente alla temperatura del sistema e ai vettori del numero di moli
iniziali e stechiometrico) questa equazione pu essere risolta nellunica incognita .
7.2.4.3 Composti in fase solida nello stato del sistema
Nei settori di interesse dellingegnere chimico si incontrano solitamente solidi puri che
reagiscono con altri composti in fase liquida o gassosa. In questo caso lo stato di
riferimento pi utilizzato composto solido puro a P
r
= 1 [bar]. Lattivit si calcola quindi
come:
( )
( ) ] r [ba 1 ,
,
=
=
P T f
P T f
a
S
i
S
i
i
(798)
Nel caso in cui la correzione di Poynting sia trascurabile la relazione si semplifica nella:
1 =
i
a (799)
che traduce la nota affermazione secondo cui lattivit dei solidi puri unitaria.
7.2.5 Influenza della temperatura sulla costante di equilibrio
Come detto in precedenza, i valori dellenergia libera di Gibbs di formazione di molti
composti sono tabulati in uno stato di riferimento standard a 298,15 [K] e 1 [bar]. Ne
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
286
consegue che immediato il calcolo del
o
R
G e quindi della costante di equilibrio a 298,15
[K] e 1 [bar]. Se si utilizzano gli stati di riferimento standard discussi in precedenza
linfluenza della pressione non importante in quanto la pressione di riferimento sempre
presa pari a 1 [bar]. Viceversa, la temperatura quella del sistema e quindi necessario
poter calcolare la costante di equilibrio a una temperatura qualsiasi partendo dal valore a
298,15 [K].
Lequazione (261) fornisce la dipendenza dellenergia libera di Gibbs molare dalla
temperatura:
( )
2
RT
h
T
RT g
i
P
i
=
(
(800)
Questa relazione pu essere rimaneggiata algebricamente per dare:
( )
( ) ( )
( )
2
2
1 1
1
2
1
RT
H
dT
RT G d
RT
h
T
RT g
RT
h
T
RT g
o
R
o
R
N
i
i i
P
N
i
i i
N
i
i
i
N
i P
i
i
=
(
(
(
(
(
|
|
.
|
\
|
|
.
|
\
|
=
(
= =
= =
(801)
La derivata parziale stata sostituita nellultima relazione dalla derivata totale in quanto il
o
R
G viene calcolato a un valore fissato di pressione, quello dello stato di riferimento. Per
analogia con il
o
R
G si introdotta la variazione dellentalpia della reazione,
o
R
H :
( ) ( )
=
=
N
i
r i i r
o
R
P T h P T H
1
, , (802)
Il significato fisico del
o
R
H analogo a quello del
o
R
G : esso indica la variazione di
entalpia associata alla trasformazione completa dei reagenti presenti in quantit
stechiometriche nei prodotti nelle condizioni di riferimento. Per esempio, il
o
R
H della
reazione (758) pari a
2 2
2 1
CO O CO
o
R
h h h H + = , cio alla variazione di entalpia che si
ha quando una mole di CO e mezza mole di O
2
si trasformano in una mole di CO
2
.
Utilizzando per il calcolo dellentalpia lo stesso stato di riferimento utilizzato per lenergia
libera di Gibbs (cio elementi chimici nella forma pi stabile nello stato di riferimento)
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
287
lentalpia nello stato di riferimento pu essere calcolata analogamente allenergia libera di
Gibbs come
181
:
( ) ( )
+ =
T
T
i P r r
o
i f r i
r
dT c P T h P T h
, ,
, ,
(803)
Come per lenergia libera di Gibbs di formazione, anche i valori dellentalpia di formazione
di molti composti sono tabulati in uno stato di riferimento standard a 298,15 [K] e 1 [bar].
La relazione (801) fornisce linfluenza della temperatura sulla costante di equilibrio:
( ) ( ) ( )
2 2
ln ln
RT
H
dT
K d
RT
H
dT
K d
dT
RT G d
o
R
o
R
o
R
=
(804)
nota come equazione di vant Hoff, da cui si vede che se una reazione esotermica
( 0 <
o
R
H ) un aumento della temperatura provoca una diminuzione della K (la reazione si
sposta verso i reagenti e quindi sfavorita da un aumento di temperatura), mentre se
endotermica ( 0 >
o
R
H ) un aumento della temperatura provoca un aumento della K (la
reazione si sposta verso i prodotti e quindi favorita da un aumento di temperatura).
Questo comportamento qualitativo noto come principio dellequilibrio mobile di vant
Hoff.
La relazione (804) pu essere integrata per fornire il valore della costante di equilibrio alla
temperatura del sistema:
=
T
T
o
R
T
T
r r
dT
RT
H
K d
2
ln
(805)
Se possibile considerare costante il
o
R
H (cosa ragionevole per piccole variazioni di
temperatura) lequazione precedente fornisce:
181
Questa relazione analoga a quelle gi viste per il calcolo dellentalpia dei composti puri. Lunica
differenza che lo stato di riferimento, dove il valore dellentalpia zero per definizione, corrisponde
agli elementi chimici nel loro stato di aggregazione pi stabile nello stato di riferimento. Quindi il
primo termine, ( )
r r
o
i f
P T h ,
,
, rappresenta semplicemente la differenza di entalpia tra il composto a
( )
r r
P T , e gli elementi. Lintegrale rappresenta invece, al solito, la variazione di entalpia da T
r
a T.
Questa formulazione pu naturalmente essere utilizzata anche per sistemi non reagenti, ma in questo
caso, poich tutti i termini dei bilanci contenenti i ( )
r r
o
i f
P T h ,
,
devono elidersi, rappresenta
uninutile complicazione.
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
288
( )
( )
|
|
.
|
\
|
|
|
.
|
\
|
r
o
R
r
T T R
H
T K
T K 1 1
ln (806)
Se invece non possibile trascurare la dipendenza del
o
R
H della temperatura bisogna
esplicitare tale dipendenza allinterno dellintegrale:
( )
( )
( )
( )
( )
=
=
|
|
|
.
|
\
|
+ =
= =
=
|
|
.
|
\
|
T
T
N
i
T
T
i P r r
o
i f i
T
T
N
i
i i
T
T
o
R
r
r r
r r
dT dT c P T h
RT
dT T h
RT
dT
RT
T H
T K
T K
1
, ,
2
1
2 2
,
1
1
ln
(807)
Noti i calori specifici in funzione della temperatura (solitamente in forma polinomiale) gli
integrali presenti in questa relazione possono essere risolti analiticamente.
7.2.6 Applicazione
Si calcoli la composizione di equilibrio di un sistema contenente etilene, acqua ed etanolo
in fase gas a 275 [C] e 35 [bar]. Inizialmente sono presenti 5 moli di acqua e 1 di etilene.
Si valuti inoltre linfluenza della riduzione della pressione a 1 [bar], della riduzione della
temperatura a 298,15 [K], dellaggiunta di 4 moli di azoto e della non idealit del gas. I dati
relativi ai composti puri nello stato standard (gas perfetto puro, 298,15 [K] e 1 [bar]) sono
riassunti nella seguente tabella.
Composto
o
f
H [J mol
-1
]
o
f
G [J mol
-1
]
Temperatura
critica [K]
Pressione
critica [bar]
Fattore
acentrico
Etilene 52510 68460 282.3 50.40 0.087
Acqua -241818 -228572 647.1 220.55 0.345
Etanolo -235100 -168490 513.9 61.48 0.645
Il vettore delle specie presenti in questo caso | |
T
OH H C O H H C
5 2 2 4 2
= S , mentre il
vettore degli atomi invece | | O H C = E . La matrice elementi specie in questo caso
quindi
(
(
(
=
1 1 0
6 2 4
2 0 2
A dove le righe sono relative agli atomi (C, H e O dallalto in basso) e
le colonne alle specie (etilene, acqua e etanolo da sinistra a destra). Il rango di questa
matrice NA = 3 mentre il numero delle specie N = 4. Quindi il numero di equazioni
stechiometriche che servono per risolvere il problema sono pari a NR = 4 3 = 1. Risulta
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
289
quindi necessario scrivere una sola reazione chimica bilanciata che comprenda tutte le
specie presenti, per esempio:
OH H C O H H C
5 2 2 4 2
= + (808)
Il vettore stechiometrico associato a questa reazione quindi il seguente:
| |
T
1 1 1 + = (809)
mentre il vettore del numero di moli :
| |
T
OH H C O H H C
n n n
5 2 2 4 2
= n (810)
e quello del numero di moli iniziali:
| |
T
0 5 1 =
o
n (811)
Il numero di moli dei vari composti in funzione del grado di avanzamento risulta quindi
pari a:
=
=
=
OH H C
O H
H C
n
n
n
5 2
2
4 2
5
1
= 6
t
n
(812)
e le frazioni molari sono:
=
6
6
5
6
1
5 2
2
4 2
OH H C
O H
H C
x
x
x
(813)
I valori del
o
R
H e
o
R
G nello stato di riferimento si calcolano facilmente dai dati
disponibili:
( ) ( )
] mol [J 8378
) 168490 ( ) 228572 ( 68460 1 , 298 1 , 298
1
1
,
=
=
= + = =
N
i
o
i f i
o
R
g G
(814)
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
290
( ) ( )
] mol [J 45792
) 235100 ( ) 241818 ( 52510 1 , 298 1 , 298
1
1
,
=
=
= + = =
N
i
o
i f i
o
R
h H
(815)
La costante di equilibrio a 298 [K] vale quindi:
( ) 42 , 29
298 314 , 8
8378
exp 298 =
|
|
.
|
\
|
= K (816)
Trascurando la dipendenza dellentalpia di reazione dalla temperatura la costante di
equilibrio a 275 [C] si pu calcolare come:
( ) ( ) 0064 , 0
298
1
548
1
314 , 8
45792
exp 298 548 =
|
|
.
|
\
|
|
.
|
\
|
= K K (817)
Anche da queste valutazioni approssimate appare evidente che linfluenza della temperatura
sulla costante di equilibrio non pu essere solitamente trascurata.
Con il riferimento utilizzato per il calcolo della costante di equilibrio, se si assume il gas
perfetto le attivit coincidono con le pressioni parziali e la relazione di equilibrio diventa:
( )( )
= = =
5 1 35
6
0064 , 0
2 4 2
5 2
O H H C
OH H C
Px Px
Px
K
(818)
Questa equazione pu essere facilmente risolta per fornire il valore di 1567 , 0 = . La
composizione del sistema nello stato di equilibrio risulta quindi pari a:
0268 , 0
8289 , 0
1443 , 0
5 2
2
4 2
=
=
=
OH H C
O H
H C
x
x
x
(819)
La conversione delletilene, , pu essere calcolata come:
=
=
=
1
) 1 ( 1
0
0
4 2
4 2 4 2
H C
H C H C
n
n n
(820)
In questo caso particolare la conversione coincide quindi col grado di avanzamento:
1566 , 0 = .
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
291
Poich la reazione avviene con una diminuzione del numero di moli una riduzione della
pressione deve abbassare la conversione delletilene. Infatti, se si riduce la pressione a 1
[bar] la relazione di equilibrio diventa:
( )( )
= = =
5 1 1
6
0064 , 0
2 4 2
5 2
O H H C
OH H C
Px Px
Px
K
(821)
che fornisce un valore di 0053 , 0 = . La composizione del sistema nello stato di equilibrio
risulta quindi pari a:
0009 , 0
8332 , 0
1659 , 0
5 2
2
4 2
=
=
=
OH H C
O H
H C
x
x
x
(822)
e la conversione si riduce a 0053 , 0 =
Laggiunta di 4 moli di azoto (che si comporta come un composto inerte) riduce la
pressione parziale dei composti e quindi agisce come una riduzione della pressione. Il
valore della costante di equilibrio non cambia, mentre cambia il numero totale di moli che
diventa
4
5
1
2
5 2
2
4 2
=
=
=
=
N
OH H C
O H
H C
n
n
n
n
=10
t
n
(823)
Le frazioni molari sono quindi in questo caso:
=
10
10
5
10
1
5 2
2
4 2
OH H C
O H
H C
x
x
x
(824)
La relazione di equilibrio diventa:
( )( )
= = =
5 1 35
10
0064 , 0
2 4 2
5 2
O H H C
OH H C
Px Px
Px
K
(825)
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
292
che fornisce un valore di 0999 , 0 = . La composizione del sistema nello stato di equilibrio
risulta quindi pari a:
0101 , 0
4950 , 0
0909 , 0
5 2
2
4 2
=
=
=
OH H C
O H
H C
x
x
x
(826)
e la conversione risulta pari a 0999 , 0 =
Se si riduce la temperatura, essendo la reazione esotermica ( 0
0
<
R
H ), la conversione di
etilene deve aumentare. Per esempio, a 298 [K] la costante di equilibrio vale 29,42 e la
relazione di equilibrio diventa:
( )( )
= = =
5 1 35
6
42 , 29
2 4 2
5 2
O H H C
OH H C
Px Px
Px
K
(827)
Questa equazione pu essere risolta per fornire il valore di 9988 , 0 = . La composizione
del sistema nello stato di equilibrio risulta quindi pari a:
1998 , 0
8000 , 0
0002 , 0
5 2
2
4 2
=
=
=
OH H C
O H
H C
x
x
x
(828)
e la conversione risulta pari a 9988 , 0 =
Se infine si considera la miscela ideale ma non il gas perfetto, la relazione di equilibrio
diventa:
( )
( ) ( )
O H O H H C H C
OH H C OH H C
Px Px
Px
K
2 2 4 2 4 2
5 2 5 2
35 , 548 35 , 548
35 , 548
0064 , 0
= =
(829)
Utilizzando lEoS PR si possono calcolare i seguenti valori dei coefficienti di fugacit dei
gas puri: ( ) 8082 , 0 35 , 548
5 2
=
OH H C
; ( ) 9697 , 0 35 , 548
4 2
=
H C
; ( ) 8721 , 0 35 , 548
2
=
O H
. La
relazione di equilibrio diventa quindi:
( )( )
= =
5 1 35
6
8721 , 0 9697 , 0
8082 , 0
0064 , 0 K (830)
Questa equazione pu essere risolta per fornire il valore di 1594 , 0 = . La composizione
del sistema nello stato di equilibrio risulta quindi pari a:
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
293
0273 , 0
8288 , 0
1439 , 0
5 2
2
4 2
=
=
=
OH H C
O H
H C
x
x
x
(831)
e la conversione risulta pari a 1594 , 0 =
7.2.7 Sistemi complessi
Quanto visto in precedenza per il caso di sistemi semplici viene facilmente generalizzato al
caso di sistemi complessi, cio caratterizzati da NR > 1. In questo caso per risolvere il
problema non pi sufficiente unequazione di equilibrio, ma necessario disporre di un
numero di equazioni pari a NR. Questo viene fatto semplicemente scrivendo un numero di
equazioni stechiometriche (cio di reazioni chimiche) pari ad NR, ciascuna caratterizzata
dal suo grado di avanzamento che rappresentano le NR incognite del problema attraverso le
quali possibile risalire al numero di moli di tutti i composti. Tali equazioni
stechiometriche devono soddisfare due vincoli: contenere tutte le specie presenti nel
sistema (tranne ovviamente quelle inerti) ed essere linearmente indipendenti
182
.
Le NR equazioni stechiometriche che vengono utilizzate hanno la forma generale:
NR k S
N
j
j jk
... 1 0
1
= =
=
(832)
dove
jk
sono i coefficienti stechiometrici della specie i nella reazione k. Si genera quindi
una matrice stechiometrica i cui elementi sono i
jk
(che degenera ovviamente nel vettore
stechiometrico nel caso di NR = 1). Per ciascuna equazione stechiometrica vale quanto
discusso per il caso di sistemi semplici. In particolare possibile definire un grado di
avanzamento per ciascuna reazione,
k
:
k
jk
jk
d
dn
= (833)
182
Esiste un metodo automatico (detto metodo di Denbigh; si veda K. Denbigh, The principles of
chemical equilibrium, Cambridge University Press, 1966) per la scrittura di NR equazioni
stechiometriche linearmente indipendenti e coinvolgenti tutte le specie presenti. Daltro canto, quando
NR non troppo elevato semplice scrivere tali equazioni anche senza lausilio di tali metodi.
Viceversa, quando NR diventa elevato solitamente il metodo del grado di avanzamento presenta dei
problemi nella risoluzione numerica del sistema di equazioni algebriche non lineari e si preferisce
quindi utilizzare altri approcci, quali quelli basati sulla minimizzazione diretta dellenergia libera di
Gibbs del sistema.
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
294
che lega la variazione di moli della specie i dovuta alla reazione k col coefficiente
stechiometrico di quella specie in quella reazione. La variazione totale del numero di moli
della specie j sar quindi:
=
=
NR
k
k jk j
d dn
1
(834)
da cui si ricava:
=
=
NR
k
k jk
o
j j
n n
1
(835)
che pu essere espressa nella forma vettoriale compatta
183
:
+ =
o
n n
(836)
Confrontando questa equazione con lespressione del bilancio materiale (729) appare
evidente che le due equazioni coincidono pur di definire il termine di generazione per
sistemi complessi come:
=
=
NR
k
k ik i
G
1
(837)
Lequazione (836) consente di calcolare il numero di moli di tutte le specie sulla base della
conoscenza del numero di moli iniziali, della matrice stechiometrica e delle NR variabili .
Tali variabili possono essere calcolate da NR relazioni dedotte dallimporre che il sistema
raggiunga le condizioni di equilibrio.
Procedendo in maniera analoga a quanto fatto per i sistemi semplici, la condizione di
minimo dellenergia libera di Gibbs conduce alla relazione:
0
1
1
1
1
1
,
=
|
|
.
|
\
|
= = =
=
=
=
=
=
N
i
ik i
NR
k
k
N
i
NR
k
k ik i
N
i
i i P T
d d dn dG (838)
183
La deduzione di questa relazione stata condotta in modo intuitivo ma pu ovviamente essere
giustificata in modo rigoroso. Il sistema di equazioni algebriche lineari (747)
o
An An = ammette
infatti la soluzione generale + =
o
n n . Da un punto di vista matematico, i parametri devono
soddisfare lequazione A = 0 cos che la equazione (747) possa essere dedotta dalla soluzione
generale + =
o
n n . Se i parametri soddisfano lequazione A = 0 questo facilmente
dimostrato moltiplicando la soluzione generale per A per ottenere:
o o
An A An An = + = .
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
295
Poich la variazione infinitesima di tutti i gradi di avanzamento arbitraria, tutti i termini
tra parentesi della sommatoria devono annullarsi e quindi la condizione di equilibrio si
riduce alle NR relazioni:
NR k
N
i
ik i
... 1 0
1
= =
=
(839)
In pratica si sono ritrovate NR equazioni identiche a quella dedotta per il caso di una sola
reazione. Procedendo esattamente come per il caso di sistemi semplici, si ottengono le NR
equazioni di equilibrio:
( ) ( )
( )
NR k
RT
P T G
P P T a K
r
o
k R
N
i
r i k
ik
... 1
,
exp , , ,
,
1
=
|
|
.
|
\
|
= =
(840)
che rappresenta un sistema di NR equazioni algebriche non lineari nelle NR incognite che
pu essere risolto. Noti i valori di , attraverso le relazioni (836) possibile calcolare il
numero di moli di tutte le specie.
Il calcolo dellattivit delle varie specie e del
o
k R
G
,
delle diverse reazioni viene condotto
esattamente come discusso per il caso di sistemi semplici.
7.2.8 Applicazione
Calcolare la composizione di equilibrio di un sistema contenente 1-propene (P), butano (B),
1,2-dimetilpentano (M) e 1-esene (E) in fase gassosa a 500 [C] e 1 [bar]. Inizialmente sono
presenti 1 mole di P e 1 mole di B.
Procedendo come mostrato nellapplicazione precedente il vettore delle specie presenti in
questo caso | |
T
E M B P = S , mentre il vettore degli atomi invece | | H C = E . In
questo caso N = 4, NA = 2 e NR = 2. Si devono quindi scrivere due reazioni chimiche
linearmente indipendenti che coinvolgano le quattro specie presenti. Per esempio si
possono utilizzare
184
:
E P
M B P
=
= +
2
(841)
184
Ovviamente anche altre reazioni che soddisfano i due vincoli detti possono essere egualmente
utilizzate conducendo allo stesso risultato.
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
296
le cui costanti di equilibrio (calcolate come illustrato nelle applicazione precedenti, con
stato di riferimento: gas perfetto puro a 500 [C] e 1 [bar]) valgono 34,5 e 7,2,
rispettivamente. La matrice stechiometrica associata a queste due reazioni :
(
(
(
(
+
+
=
1 0
0 1
0 1
2 1
(842)
mentre il vettore del numero di moli :
| |
T
E M B P
n n n n = n (843)
e quello del numero di moli iniziali:
| |
T
0 0 1 1 =
o
n (844)
Il numero di moli dei vari composti in funzione dei gradi di avanzamento risulta quindi pari
a:
2
1
1
2 1
1
2 1
=
=
=
=
E
M
B
P
n
n
n
n
2 1
2 =
t
n
(845)
e le frazioni molari sono:
2 1
2
2 1
1
2 1
1
2 1
2 1
2
2
2
1
2
2 1
=
=
=
=
E
M
B
P
x
x
x
x
(846)
Con il riferimento utilizzato per la costante di equilibrio, se si assume il gas perfetto le
attivit coincidono con le pressioni parziali e le relazioni di equilibrio diventano:
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
297
( )( )
( ) ( )
2
2 1
2 2 1
2
2
1 2 1
1 2 1
1
2 1
1
2
2 , 7
1 2 1 1
2
5 , 34
= = =
= = =
P
E
B P
M
Px
Px
K
Px Px
Px
K
(847)
Questo sistema di equazioni algebriche non lineari pu essere risolto numericamente
185
per
fornire il valore di
T
] 042 , 0 789 , 0 [ = .
7.3 Sistemi aperti in condizioni stazionarie
La trattazione dei sistemi aperti in condizioni stazionarie strettamente analoga a quella
sviluppata per i sistemi chiusi. Tale stretta analogia facilmente intuibile confrontando le
equazioni di bilancio materiale per un composto nei due casi. Lequazione (729), valida per
sistemi chiusi, :
i
o
i i
G n n = (848)
mentre lequazione (733), valida per sistemi aperti in condizioni stazionarie, :
i IN i OUT i
G n n
&
& & =
, ,
(849)
Si vede che le due equazioni sono matematicamente identiche. Cambia solo il significato
dei simboli, che per sistemi chiusi rappresentano il numero di moli, mentre per sistemi
aperti le portate molari. Entrambe le equazioni fanno inoltre riferimento a due stati: quello
iniziale e quello finale (di equilibrio) nel caso di sistemi chiusi, quello in ingresso e quello
in uscita (di equilibrio) nel caso di sistemi aperti in condizioni stazionarie.
Per rimarcare questa analogia nel seguito si indicheranno con lapice
0
le condizioni in
ingresso, e senza apice quelle in uscita. la relazione precedente diventa quindi, con questa
notazione:
i i i
G n n
&
& & =
0
(850)
Lanalogia completa anche per quanto riguarda il calcolo delle frazioni molari: nel caso di
sistemi chiusi vengono calcolate come rapporto tra il numero di moli di un composto e il
numero di moli totali (sempre riferite a una data fase),
=
=
N
j
j i i
n n x
1
, mentre nel caso
delle correnti che entrano o escono da un sistema aperto vengono calcolate come rapporto
tra la portata molare di un composto e la portata molare totale (sempre riferite a una data
corrente),
=
=
N
j
j i i
n n x
1
& & .
185
Per esempio col comando fsolve di MATLAB.
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
298
Anche la condizione di equilibrio la stessa: la composizione del sistema nello stato di
equilibrio (quello finale per i sistemi chiusi o quello in uscita per i sistemi aperti in
condizioni stazionarie) deve essere tale da rendere minima lenergia libera di Gibbs
186
alla
temperatura e pressione assegnata.
Procedendo quindi in maniera assolutamente analoga a quella discussa per il caso di sistemi
chiusi
187
, si ricavano le equazioni che consentono di calcolare la composizione della
corrente uscente in condizioni di equilibrio. Queste relazioni sono riassunte di seguito per il
caso generale di sistemi complessi. Le relazioni per sistemi semplici derivano ovviamente
da queste con NR = 1.
Per risolvere il problema sempre necessario scrivere NR equazioni stechiometriche:
NR k S
N
j
j jk
... 1 0
1
= =
=
(851)
Per ciascuna equazione stechiometrica possibile definire un grado di avanzamento,
k
&
188
, che lega la variazione della portata molare del singolo composto:
=
=
NR
k
k jk j
d n d
1
&
& (852)
da cui si ricava, integrando tra le condizioni in ingresso e quelle in uscita:
=
=
NR
k
k jk
o
j j
n n
1
&
& & (853)
che pu essere espressa nella forma vettoriale compatta:
&
& & + =
o
n n
(854)
Confrontando questa equazione con lespressione del bilancio materiale (729) appare
evidente che le due equazioni coincidono pur di definire il termine di generazione come:
=
=
NR
k
k jk i
G
1
& &
(855)
Anche in questo caso, lequazione (853) non rappresenta altro che una forma del bilancio
materiale per la specie j in condizioni stazionarie che include implicitamente il vincolo
stechiometrico, cio il vincolo di conservazione delle specie atomiche.
186
Nel caso di sitemi aperti si tratta dellenergia libera di Gibbs associata alla portata uscente, e
quindi di un flusso di energia libera di Gibbs.
187
Si lascia allo studente per esercizio la derivazione di dettaglio.
188
Si noti che ovviamente in questo caso il grado di avanzamento non ha dimensioni [mol] come per
il caso dei sistemi chiusi, ma bens [mol s
-1
] poich correla la variazione di portate molari e non pi di
numero di moli.
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
299
Queste equazioni consentono di calcolare la portata molare di tutte le specie sulla base della
conoscenza delle portate molari in ingresso, della matrice stechiometrica e delle NR
variabili
k
&
. Tali variabili possono essere calcolate da NR relazioni dedotte dallimporre
che la corrente in uscita raggiunga le condizioni di equilibrio. Questo porta alle stesse NR
relazioni dedotte per il caso di sistemi chiusi:
( )
( )
NR k
RT
P T G
P P T a K
r
o
k R
N
i
r i k
ik
... 1
,
exp , , ,
,
1
=
|
|
.
|
\
|
= |
.
|
\
|
=
&
(856)
che rappresenta un sistema di NR equazioni algebriche non lineari nelle NR incognite
&
che pu essere risolto. Noti i valori di
&
, attraverso le relazioni (854) possibile calcolare
le portate molari di tutte le specie.
Il calcolo dellattivit delle varie specie e del
o
k R
G
,
delle diverse reazioni viene condotto
esattamente come discusso per il caso di sistemi chiusi.
7.3.1 Applicazione
Il butadiene pu essere preparato per deidrogenazione catalitica dell1-butene a 900 [C] e
1 [bar]. Per eliminare le reazioni secondarie si pu diluire lalimentazione di 1-butene con
vapor dacqua nel rapporto 1-butene/acqua di 1/10. Calcolare la composizione di equilibrio
della corrente uscente dal reattore.
Procedendo come mostrato nellapplicazione precedente il vettore delle specie presenti in
questo caso | |
T
O H H H C H C
2 2 6 4 8 4
= S , mentre il vettore degli atomi invece
| | O H C = E . In questo caso N = 4, NA = 3 e NR = 1. Si deve quindi scrivere una
reazione chimica che coinvolga le specie presenti eccetto lacqua, in quanto (poich
lossigeno presente solo in questa molecola) essa non pu trasformarsi in nessun altra
molecola. In altri termini, lacqua agisce come un diluente inerte.
Per esempio si pu utilizzare:
2 6 4 8 4
H H C H C + = (857)
la cui costante di equilibrio (calcolata come illustrato nelle applicazione precedenti, con
stato di riferimento: gas perfetto puro a 900 [C] e 1 [bar]) vale 0,242. Il vettore
stechiometrico associato a questa reazione :
| | 0 1 1 1 = (858)
mentre il vettore delle portate molari :
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
300
| |
T
O H H H C H C
n n n n
2 2 6 4 8 4
& & & & & = n (859)
e quello delle portate molari entranti (scegliendo come base di calcolo arbitraria, in quanto
non influenza i risultati in termini di composizione della corrente uscente, 1 [mol s
-1
] di 1-
butene):
| |
T
10 0 0 1 =
o
n& (860)
Le portate molari dei vari composti in funzione del grado di avanzamento risultano quindi
pari a:
10
1
2
2
6 4
8 4
=
=
=
=
O H
H
H C
H C
n
n
n
n
&
&
&
&
&
&
&
&
& + =11
t
n
(861)
e le frazioni molari sono:
&
&
&
&
&
&
&
+
=
+
=
+
=
+
=
11
10
11
11
11
1
2
2
6 4
8 4
O H
H
H C
H C
x
x
x
x
(862)
Con il riferimento utilizzato per la costante di equilibrio, se si assume il gas perfetto le
attivit coincidono con le pressioni parziali e la relazione di equilibrio diventa:
1
2
1 11
1
242 , 0
8 4
2 6 4
&
&
&
+
= = =
H C
H H C
Px
Px Px
K
(863)
Questa equazione algebrica pu essere risolta per fornire il valore di 784 , 0 =
&
. Le frazioni
molari della corrente uscente sono quindi:
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
301
8486 , 0
0665 , 0
0665 , 0
0184 , 0
2
2
6 4
8 4
=
=
=
=
O H
H
H C
H C
x
x
x
x
(864)
7.4 Bilancio di energia in sistemi multicomponente
Il bilancio di energia per sistemi multicomponente identico a quello discusso per il caso
monocomponente. Lunica differenza risiede nel calcolo dei valori delle grandezze
termodinamiche (entalpia ed energia interna specifica) che vanno effettuate coi metodi
discussi per le miscele e con la scelta dello stato di riferimento opportuno (cio, in presenza
di sistemi reagenti, elementi chimici nella forma pi stabile nello stato di riferimento). In
particolare, importante sottolineare che anche per sistemi reagenti (cio anche in presenza
di reazioni esotermiche o endotermiche) il termine di generazione sempre nullo. Il
riscaldamento o raffreddamento della miscela a seguito delle trasformazioni chimiche viene
automaticamente tenuto in conto nei bilanci grazie alla scelta di uno stato di riferimento
comune a tutte le specie.
Il bilancio di energia semplificato trascurando i termini di energia cinetica e potenziale
assume quindi la forma:
( )
W Q h n h n
dt
nu d
OUT OUT IN IN
& &
& & + + = (865)
Per sistemi non reagenti lapplicazione dellequazione di bilancio identica al caso
monocomponente pur di calcolare i valori dellentalpia e dellenergia interna specifica con
le relazioni discusse per le miscele.
Per sistemi reagenti invece, analogamente al caso dei bilanci materiali, nellambito della
termodinamica si possono utilizzare due forme semplificate dei bilanci di energia per i
sistemi reagenti: quello per sistemi chiusi (in cui si assume che lo stato finale sia di
equilibrio) e quello in condizioni stazionarie (in cui si assume che lo stato della corrente in
uscita sia di equilibrio). Per semplicit di trattazione si considerano inoltre nulle nel seguito
le potenze termiche e meccaniche scambiate dal sistema con lambiente: il considerarle non
nulle complica la scrittura delle equazioni senza modificarne lo sviluppo.
7.4.1 Sistemi chiusi
Lequazione di bilancio di energia in questo caso si riduce alla seguente relazione, in cui
non compaiono i termini di scambio di energia con lambiente:
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
302
( )
( )
( )
nu u n nu d
dt
nu d
o o
nu
nu
o
= = =
0 0
(866)
che, in termini di entalpia, diventa:
( ) ( ) Pv h n v P h n
o o o o
= (867)
Per trasformazioni a pressione e volume costante, o comunque in tutti i casi in cui il
termine Pv risulta trascurabile rispetto al termine entalpico
189
, questa relazione pu essere
approssimata dalla seguente:
nh h n
o o
=
(868)
Lentalpia molare di una miscela si calcola come media pesata sulle frazioni molari delle
entalpie parziali molari. Nel caso in cui la miscela si comporti idealmente queste
coincidono con le entalpie molari per cui, trascurando anche la dipendenza dellentalpia
molare dalla pressione (per cui non si riporta in modo esplicito nelle variabili la dipendenza
dalla pressione), si ottiene:
( ) ( ) ( )
= =
|
|
|
.
|
\
|
+ = =
N
i
T
T
i P r
o
i f i
N
i
i i
r
dT c T h x T h x T h
1
, ,
1
(869)
Introducendo questa relazione nellequazione di bilancio:
( ) ( )
= =
|
|
|
.
|
\
|
+ =
|
|
|
.
|
\
|
+
N
i
T
T
i P r
o
i f i
N
i
T
T
i P r
o
i f i
o
r
o
r
dT c T h x n dT c T h x n
1
, ,
1
, ,
(870)
e portando il numero di moli allinterno della sommatoria si ottiene:
( ) ( )
= =
|
|
|
.
|
\
|
+ =
|
|
|
.
|
\
|
+
N
i
T
T
i P r
o
i f i
N
i
T
T
i P r
o
i f
o
i
r
o
r
dT c T h n dT c T h n
1
, ,
1
, ,
(871)
Introducendo in questa equazione il bilancio di materia (835) e spezzando lintegrale al
secondo membro si ottiene:
189
Come normalmente avviene per le fasi condensate.
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
303
( )
( )
=
=
=
|
|
|
.
|
\
|
+ + |
.
|
\
|
+ =
=
|
|
|
.
|
\
|
+
N
i
T
T
i P
T
T
i P r
o
i f
NR
k
k ik
o
i
N
i
T
T
i P r
o
i f
o
i
o
o
r
o
r
dT c dT c T h n
dT c T h n
1
, , ,
1
1
, ,
(872)
Semplificando i termini uguali, lequazione precedente si riduce alla:
( ) 0
1
, ,
1
1
,
=
|
|
|
.
|
\
|
+ +
=
=
=
N
i
T
T
i P r
o
i f
NR
k
k ik
N
i
T
T
i P
o
i
r
o
dT c T h dT c n (873)
Lultimo termine tra parentesi non altro che il ( ) ( )
+ =
T
T
i P r
o
i f
o
i f
r
dT c T h T h
, , ,
:
( ) 0
1
,
1 1
,
=
|
|
.
|
\
|
+
= = =
NR
k
o
i f
N
i
ik k
N
i
T
T
i P
o
i
T h dT c n
o
(874)
mentre lultimo termine tra parentesi non altro che il ( ) ( ) T h T H
o
i f
N
i
ik
o
k R ,
1
,
=
=
, per cui
la relazione precedente diviene:
( )
= =
=
NR
k
o
k R k
N
i
T
T
i P
o
i
T H dT c n
o
1
,
1
,
(875)
Questi due termini hanno un evidente significato fisico: quello a sinistra rappresenta
lenergia necessaria per riscaldare (o raffreddare) i reagenti dalla temperatura iniziale, T, a
quella finale, T. Il termine a destra rappresenta la somma dellenergia liberata (o assorbita)
da ciascuna reazione se avvenisse completamente partendo da reagenti stechiometrici alla
temperatura di equilibrio, il ( ) T H
o
k R,
, pesata col proprio grado di avanzamento. In altri
termini, si tratta dellenergia liberata (o assorbita) dalle trasformazioni chimiche che
avvengono alla temperatura di equilibrio T. Se il sistema non scambia energia con
lambiente (come in questo caso) evidente che i due termini devono bilanciarsi, cio
lenergia liberata (o assorbita) dalle trasformazioni chimiche serve a riscaldare (o
raffreddare) i reagenti.
Se il sistema scambia potenza termica o potenza meccanica con lambiente questi
compariranno ovviamente nella relazione finale andando a sbilanciare luguaglianza tra
energia liberata (o assorbita) dalle trasformazioni chimiche e quella necessaria per
riscaldare (o raffreddare) i reagenti.
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
304
La relazione precedente si semplifica notevolmente per il caso di NR = 1 se si assume
cost
o
R
H e si considera un calore specifico massico di miscela anchesso costante. In
questo caso, dovendosi conservare la massa totale presente nel sistema,
( )
o
mix P
o
N
i
T
T
i P
o
i
T T c m dT c n
o
=
,
1
,
(876)
da cui si deduce la relazione approssimata:
mix P
o
o
R
ad
o
c m
H
T T T
,
=
(877)
che consente una stima rapida del cosiddetto incremento adiabatico di temperatura,
ad
T
190
, cio del massimo incremento di temperatura possibile della massa iniziale del
sistema a causa delle reazioni esotermiche.
7.4.2 Sistemi aperti in condizioni stazionarie
Lequazione di bilancio di energia in questo caso si riduce alla seguente relazione:
OUT OUT IN IN
h n h n & & = (878)
Si vede immediatamente che questa equazione matematicamente identica alla equazione
(868) da cui si dedotta la (875). Cambia solo il significato dei simboli, che per sistemi
chiusi rappresentano il numero di moli, mentre per sistemi aperti le portate molari.
Entrambe le equazioni fanno inoltre riferimento a due stati: quello iniziale e quello finale
(di equilibrio) nel caso di sistemi chiusi, quello in ingresso e quello in uscita (di equilibrio)
nel caso di sistemi aperti in condizioni stazionarie.
Ne consegue che dalla relazione (878) si pu dedurre, con passaggi del tutto analoghi a
quelli visti per sistemi chiusi e utilizzando il bilancio materiale (853) una relazione analoga
alla (875):
( )
= =
=
NR
k
OUT
o
k R k
N
i
T
T
i P IN i
T H dT c n
OUT
IN
1
,
1
, ,
&
& (879)
190
Questo parametro gioca un ruolo rilevante per esempio nella stima della massima temperatura che
pu essere raggiunta in un reattore chimico nel corso di un evento incidentale dovuto alla rottura del
sistema di raffreddamento del reattore stesso.
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
305
Inserendo lapice
0
per le condizioni in ingresso e nessun apice per quelle (di equilibrio) in
uscita lanalogia formale con la relazione derivata per i sistemi chiusi diviene ancora pi
evidente:
( )
= =
=
NR
k
o
k R k
N
i
T
T
i P i
T H dT c n
1
,
1
,
0
0
&
& (880)
Anche in questo caso i due termini dellequazione hanno un evidente significato fisico:
quello a sinistra rappresenta lenergia necessaria per riscaldare (o raffreddare) la portata
entrante dalla temperatura di ingresso, T
0
, a quella in uscita, T. Il termine a destra
rappresenta la somma dellenergia liberata (o assorbita) da ciascuna reazione se avvenisse
completamente partendo da reagenti stechiometrici alla temperatura di equilibrio,
( ) T H
o
k R,
, pesata col proprio grado di avanzamento,
k
&
. In altri termini, si tratta
dellenergia liberata (o assorbita) dalle trasformazioni chimiche che avvengono alla
temperatura di equilibrio, T. Se il sistema non scambia energia con lambiente (come in
questo caso) evidente che i due termini devono bilanciarsi, cio lenergia liberata (o
assorbita) dalle trasformazioni chimiche serve a riscaldare (o raffreddare) la portata
entrante.
Analogamente al caso di sistemi chiusi, anche questa relazione si semplifica per il caso di
NR = 1 se si assume cost
o
R
H e si considera un calore specifico massico di miscela
anchesso costante. In questo caso si deduce la relazione approssimata:
mix P
o
R
ad
o
c m
H
T T T
,
0
.
&
&
=
(881)
Si tratta anche in questo caso del massimo incremento di temperatura della portata entrante
a causa delle reazioni esotermiche.
7.4.3 Applicazione
Si calcoli la composizione e la temperatura della corrente uscente (in equilibrio) da un
reattore adiabatico di sintesi dellammoniaca da idrogeno e azoto nellipotesi che i gas si
comportino come un gas perfetto. La portata entrante costituita da una miscela
stechiometrica al 25% in moli di azoto e 75% di idrogeno a 25 [C] e 100 [bar]. I parametri
necessari ai calcoli (c
P
/ R = a + b T + c T
-2
) sono riportati nella tabella seguente.
Composto ) 298 (
o
f
h [J mol
-1
] ) 298 (
o
f
g [J mol
-1
] a b c
Azoto 0 0 3,280 0,593 0,040
Idrogeno 0 0 3,249 0,422 0,083
Ammoniaca - 45940 - 16401 3,578 3,020 -0,816
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
306
Procedendo come mostrato nelle applicazioni precedenti il vettore delle specie presenti in
questo caso | |
T
NH H N
3 2 2
= S , mentre il vettore degli atomi invece | | H N = E .
In questo caso N = 3, NA = 2 e NR = 1. Si deve quindi scrivere una reazione chimica che
coinvolga tutte le specie presenti. Per esempio si pu utilizzare:
3 2 2
2
3
2
1
NH H N = + (882)
Il vettore stechiometrico associato a questa reazione :
| |
T
1 2 / 3 2 / 1 = (883)
mentre il vettore delle portate molari :
| |
T
NH H N
n n n
3 2 2
& & & & = n (884)
e quello delle portate molari entranti (scegliendo come base di calcolo, che non influenza i
risultati in termini di composizione della corrente uscente, una portata molare totale di 1
[mol s
-1
]):
| |
T
0 75 , 0 25 , 0 =
o
n& (885)
Le portate molari dei vari composti in funzione del grado di avanzamento risultano quindi
pari a:
&
&
&
&
&
=
=
=
3
2
2
2
3
75 , 0
2
1
25 , 0
NH
H
N
n
n
n
&
=1
t
n
(886)
e le frazioni molari sono:
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
307
&
&
&
&
&
&
=
1
1
2
3
75 , 0
1
2
1
25 , 0
3
2
2
NH
H
N
x
x
x
(887)
Il problema presenta due incognite: il grado di avanzamento e la temperatura.
Come primo approccio possibile scrivere le due equazioni (di equilibrio e di bilancio di
energia) approssimate assumendo cost
o
R
H e considerando un calore specifico massico
di miscela anchesso costante.
In questo caso lequazione di bilancio di energia diventa:
0
,
=
+
mix P
o
R o
c m
H
T T
&
&
(888)
dove:
298 =
o
T [K];
45940 ) 1 , 298 ( ) 298 (
3
1
0
=
= i
fi i
o
R
h H [J mol
-1
];
5 , 8
3
1
= =
= i
i
o
i
PM n m & & [g s
-1
];
( )
7326 , 5
3
3
1
2
,
=
|
.
|
\
|
+ +
=
=
i i
o
i
o
i i
mix P
PM
R
T c T b a
c [J g
-1
].
La relazione di equilibrio, assumendo come stato di riferimento il gas perfetto puro alla T
del sistema e 1 [bar], assume la forma (in questo caso le attivit coincidono con le pressioni
parziali se si considerano, per semplicit, i gas perfetti
191
):
191
Si lascia allo studente, come esercizio, la discussione della correttezza di questa approssimazione
in questo caso particolare.
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
308
( )
( ) ( )
2 / 3 2 / 1
2 2
3
exp ) (
H N
NH
o
R
Px Px
Px
RT
T G
T K =
|
|
.
|
\
|
=
(889)
Le frazioni molari si possono esprimere in funzione del grado di avanzamento come
mostrato sopra, mentre la dipendenza approssimata della costante di equilibrio della
temperatura risulta nella:
( ) ( )
|
|
.
|
\
|
|
.
|
\
|
=
298
1 1
314 , 8
45940
exp 298
T
K T K (890)
Il valore della costante di equilibrio a 298 [K] si calcola facilmente come:
( ) 9 , 749
298 314 , 8
3 , 16401
exp 298 =
|
|
.
|
\
|
= K (891)
essendo, analogamente al ) 298 (
o
R
H :
( ) ( )
] mol [J 3 , 16401
) 3 , 16401 ( 0
2
3
0
2
1
1 , 298 1 , 298
1
1
,
=
=
= + = =
N
i
o
i f i
o
R
g G
(892)
Il sistema di due equazioni algebriche non lineari (888) e (892) pu essere risolto
numericamente a dare: 0,3293 =
&
[mol s
-1
] e T = 608,5 [K]. Le frazioni molari della
corrente uscente risultano quindi pari a x = [0,1273 0,3818 0,4910]
T
.
Il sistema pu essere risolto con un programma di calcolo in linguaggio MATLAB, come al
solito riportato di seguito commentato e quindi di facile lettura. Questo programma pu
essere implementato in un file testo di nome reaz.m.
%reaz calcolo dell'equilibrio adiabatico in sistemi reagenti
% reattore in flusso gassoso con 1/2 N2 + 3/2 H2 = NH3
% approccio semplificato
%
% R. Rota (2002)
clear all
close all
format short
R = 8.314; %cost gas [J / (mol K)]
% sistema [N2 H2 NH3]': dati
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
309
DHf = [0 0 -45940]'; %DHo formazione [J/mol]
DGf = [0 0 -16401.3]'; %DGo formazione [J/mol]
% cp*/R = a+bT+cT^(-2) T in [K]
a = [3.280 3.249 3.578]'*R;
b = [0.593 0.422 3.020]'./1e3*R;
c = [0.040 0.083 -0.816]'*1e5*R;
nu= [-1/2 -3/2 1]'; %vettore stechiometrico
PM= [28 2 17]'; %vettore pesi molecolari
no= [0.25 0.75 0]'; %vettore portate IN, [mol/s]
xo= no./sum(no); %vettore frazioni molari IN
P = 100; %pressione [bar]
To = 500; %temperatura IN [K]
DHo = DHf'*nu; %DHo(298) [J/mol]
DGo = DGf'*nu; %DGo(298) [J/mol]
Ko = exp(-DGo/(R*298)); %Keq(298)
% stima con DHo e cp costanti
cpo = mean((a + b*To + c*To^(-2))./PM); %stima di cpmix [J/g]
mo = sum(no.*PM); %portata IN [g/s]
X = [0.25 700]; %valori di primo tentativo
global DHo mo cpo Ko R To no nu P
lT = fsolve(@freaz1,X); %risolvo il sistema
% visualizzazione dei risultati
lambda = lT(1)
T = lT(2)
n = [no + nu*lambda]'
x = n/sum(n)
Il programma reaz.m chiama un sottoprogramma, freaz1.m in cui si calcolano le
funzioni da azzerare, riportato di seguito.
function F = freaz1(X)
%freaz1 calcola i residui del bilancio di energia
% nella forma:
% T = TIN - lambda*DHo / (m*cp)
% accoppiato con la relazione di equilibrio
%
% R. Rota (2002)
global DHo mo cpo Ko R To no nu P
l = X(1);
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
310
T = X(2);
n = no + nu*l;
x = n/sum(n);
Pi=P*x;
KT= Ko*exp(-DHo/R*(1/T - 1/298));
F(1) = T - (To-l*DHo / (mo*cpo));
F(2) = KT - prod(Pi.^nu);
Questi valori possono essere utilizzati come primo tentativo per la risoluzione del sistema
di due equazioni pi complesso che risulta se non si considerano le approssimazioni
utilizzate in precedenza. In questo caso il bilancio di energia diventa:
( ) ( ) 0
3
1
298
2
,
= + + +
T H dT T c T b a n
o
R
i
T
i i i IN i
&
& (893)
dove lintegrale pu ovviamente essere risolto analiticamente fornendo una funzione della
temperatura incognita, T. Analogamente si pu calcolare laltra funzione della temperatura
incognita, ( ) ( ) ( )
=
+ + + =
T
i i i
i
i
o
R
o
R
dT T c T b a H T H
298
2
3
1
298 .
La dipendenza della costante di equilibrio dalla temperatura si complica anchessa
diventando:
( ) ( )
( ) ( )
|
|
|
|
|
.
|
\
|
+ + +
=
=
T
T
i i i
i
i
o
R
dT
RT
dT T c T b a H
K T K
298
2
298
2
3
1
298
exp 298
(894)
Anche in questo caso entrambi gli integrali presenti possono essere risolti analiticamente
fornendo una ulteriore funzione della temperatura incognita
192
che pu essere inserita nella
relazione di equilibrio:
( ) ( )
2 / 3 2 / 1
2 2
3
) (
H N
NH
Px Px
Px
T K =
(895)
192
Si noti che la risoluzione dellintegrale pi interno non fornisce un numero, ma bens un funzione
di T che a sua volta diventa largomento dellintegrale pi esterno e deve quindi essere nuovamente
integrata.
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
311
Il sistema di due equazioni algebriche non lineari (893) e (895) pu essere risolto
numericamente a dare: 0,1998 =
&
[mol s
-1
] e T = 661,9 [K]. Le frazioni molari della
corrente uscente risultano quindi pari a x = [0,1876 0,5628 0,2496]
T
.
Il sistema pu essere risolto estendendo il programma di calcolo in linguaggio MATLAB gi
illustrato in precedenza, come al solito riportato di seguito commentato e quindi di facile
lettura.
%reaz calcolo dell'equilibrio adiabatico in sistemi reagenti
% reattore in flusso gassoso con 1/2 N2 + 3/2 H2 = NH3
%
% R. Rota (2002)
clear all
close all
format short
R = 8.314; %cost gas [J / (mol K)]
% sistema [N2 H2 NH3]': dati
DHf = [0 0 -45940]'; %DHo formazione [J/mol]
DGf = [0 0 -16401.3]'; %DGo formazione [J/mol]
% cp*/R = a+bT+cT^(-2) T in [K]
a = [3.280 3.249 3.578]'*R;
b = [0.593 0.422 3.020]'./1e3*R;
c = [0.040 0.083 -0.816]'*1e5*R;
nu= [-1/2 -3/2 1]'; %vettore stechiometrico
PM= [28 2 17]'; %vettore pesi molecolari
no= [0.25 0.75 0]'; %vettore portate IN, [mol/s]
xo= no./sum(no); %vettore frazioni molari IN
P = 100; %pressione [bar]
To = 500; %temperatura IN [K]
DHo = DHf'*nu; %DHo(298) [J/mol]
DGo = DGf'*nu; %DGo(298) [J/mol]
Ko = exp(-DGo/(R*298)); %Keq(298)
% stima con DHo e cp costanti
cpo = mean((a + b*To + c*To^(-2))./PM); %stima di cpmix [J/g]
mo = sum(no.*PM); %portata IN [g/s]
X = [0.25 700]; %valori di primo tentativo
global DHo mo cpo Ko R To no nu P
lT = fsolve(@freaz1,X); %risolvo il sistema
% visualizzazione dei risultati
lambda = lT(1)
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
312
T = lT(2)
n = [no + nu*lambda]'
x = n/sum(n)
pause
% stima con DHo e cp variabili
% sommatoria nui*ai, nui*bi, nui*ci
am = a'*nu;
bm = b'*nu;
cm = c'*nu;
% sommatoria ai*niIN, bi**niIN, ci*niIN
aIN= a'*no;
bIN= b'*no;
cIN= c'*no;
%dichiarazione di variabili simboliche
syms t1 t2 T DHTs KTs HINTs
% calcolo delle funzioni di T con integrazione simbolica
DHTs = DHo+int(am+bm*t2+cm*t2^(-2),t2,298,T);
HINTs= int(aIN+bIN*t2+cIN*t2^(-2),t2,To,T);
KTs = Ko*exp(int((DHo+int(am+bm*t2+cm*t2^(-
2),t2,298,t1))./(R*t1^2),t1,298,T));
X = lT; %valori di primo tentativo
global no nu P DHTs KTs HINTs
lT = fsolve(@freaz2,X); %risolvo il sistema
% visualizzazione dei risultati
lambda = lT(1)
T = lT(2)
n = [no + nu*lambda]'
x = n/sum(n)
Il programma reaz.m chiama due sottoprogrammi, freaz1.m, gi mostrato in precedenza,
e freaz2.m riportato di seguito.
function F = freaz2(X)
%freaz1 calcola i residui del bilancio di energia
% accoppiato con la relazione di equilibrio
%
% R. Rota (2002)
global no nu P DHTs KTs HINTs
l = X(1);
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
313
T = X(2);
n = no + nu*l;
x = n/sum(n);
Pi=P*x;
KT=eval(KTs); %valuto la funzione K(T) alla T corrente
DHT=eval(DHTs); %valuto la funzione DHo(T) alla T corrente
HINT=eval(HINTs); %valuto la funzione HINT(T) alla T corrente
F(1) = HINT + l*DHT;
F(2) = KT - prod(Pi.^nu);
Si noti che si sfruttata la presenza di integrali tediosi da risolvere analiticamente per
mostrare le potenzialit di MATLAB nella manipolazione di espressioni simboliche. Il
comando int() non fornisce come risultato un numero, ma una espressione in cui
compare la variabile T. Questa espressione viene poi valutata a un valore corrente della
variabile T attraverso il comando eval().
Con questo programma facile ripetere i calcoli per investigare leffetto di diverse
condizioni, come riportato nella tabella seguente considerando a titolo di esempio la
pressione e la temperatura di ingresso
193
.
T [K] P [bar]
&
[mol s
-1
] T [K]
298 100 0,1998 661,9
298 1 0,1062 481,9
298 250 0,2118 685,8
400 100 0,1612 694,6
500 100 0,1158 711,2
7.5 Esercizi
Un sistema chiuso contiene CO, CO
2
, H
2
O e H
2
in fase gassosa. Calcolare la percentuale di
vapore convertita in condizioni di equilibrio se inizialmente sono presenti 1 mole di CO e 1
mole di H
2
O e il sistema viene mantenuto a 1100 [K] e 1 [bar]. Si consideri il gas perfetto.
Si valuti inoltre leffetto dellaumento della pressione a 10 [bar].
Risultato: 50 %; nessun effetto.
Un sistema chiuso contiene CO, CO
2
, H
2
O, H
2
e N
2
in fase gassosa. Calcolare la
percentuale di vapore convertita in condizioni di equilibrio se inizialmente sono presenti 1
193
Viene lasciato per esercizio allo studente il commento di questi risultati: era possibile prevedere
qualitativamente linfluenza di temperatura e pressione su
&
?
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
314
mole di CO, 1 mole di H
2
O e 2 moli di N
2
e il sistema viene mantenuto a 1100 [K] e 1
[bar]. Si consideri il gas perfetto. Si valuti inoltre leffetto dellaumento della temperatura a
1650 [K].
Risultato: 50 %; 36%.
Un sistema chiuso contiene CO, CO
2
, H
2
O e H
2
in fase gassosa. Calcolare la percentuale di
vapore convertita in condizioni di equilibrio se inizialmente sono presenti 1 mole di CO e 2
moli di H
2
O, oppure 2 moli di CO e 1 mole di H
2
O e il sistema viene mantenuto a 1100 [K]
e 1 [bar]. Si consideri il gas perfetto.
Risultato: 33 %; 66%.
La reazione SO
2
+ 0,5 O
2
= SO
3
viene condotta in fase gas a 800 [K] e 1 [bar] fino al
raggiungimento delle condizioni di equilibrio. La costante di equilibrio, calcolata
assumendo come riferimento i composti puri come gas perfetti alla temperatura del sistema
e alla pressione di 1 [bar], vale 5,99. Considerando i gas perfetti, si ottiene una conversione
dellanidride solforosa maggiore partendo da una miscela composta da 20 moli di SO
2
e 80
di O
2
o da una miscela composta da 2 moli SO
2
, 8 di O
2
e 90 di N
2
?
Risultato: dalla miscela 20 / 80.
A un reattore di isomerizzazione si alimenta una miscela equimolare in fase vapore dei tre
xileni isomeri. Se il reattore lavora a 700 [K] e 1 [bar], quale la composizione della
miscela in uscita? Si assumano i gas perfetti e la corrente uscente in condizioni di
equilibrio. I valori dei
o
f
g dei tre composti come gas perfetti puri a 700 [K] e 1 [bar]
sono: 64500, 65510 e 65560 [cal mol
-1
] per meta-, para- e orto-xilene, rispettivamente.
Risultato: y
orto
= 0,513; y
para
= 0,248.
Calcolare la minima portata di azoto da aggiungere a una miscela stechiometrica idrogeno
aria alimentata a un reattore adiabatico perch la temperatura della corrente in uscita, nelle
condizioni di equilibrio, non superi i 700 [K]. La portata alimentata contiene 1 [mol s
-1
] di
idrogeno e si trova a 298 [K] e 1 [bar]. Si assumano i gas perfetti.
Risultato: 19 [mol s
-1
].
Una sistema chiuso contenente inizialmente 10 moli di azoto e 10 moli di idrogeno viene
mantenuto a 450 [K] e 4 [bar]. In queste condizioni, in presenza di opportuni catalizzatori,
si forma anche ammoniaca. Assumendo i gas perfetti si calcoli la composizione della
miscela in condizioni di equilibrio.
Risultato: y
N2
= 0,5; y
H2
= 0,3.
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
315
Una sistema chiuso contenente inizialmente 2 moli di metano e 3 moli di acqua viene
mantenuto a 1000 [K] e 1 [bar]. In queste condizioni si formano anche monossido di
carbonio, anidride carbonica e idrogeno. Assumendo i gas perfetti si calcoli la
composizione della miscela in condizioni di equilibrio.
Risultato: y
CH4
= 0,0205; y
CO
= 0,1735; y
CO2
= 0,0374; y
H2O
= 0,0988
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
317
8 Appendice A
Richiami di matematica
In questa Appendice vengono brevemente riassunti i principali strumenti matematici
necessari alla comprensione del materiale esposto nel testo.
8.1 Relazioni fondamentali
8.1.1 Logaritmi ed esponenziali
b a
y
e e b a
a y a
b a
b
a
b a ab
= +
=
= |
.
|
\
|
+ =
) exp(
ln ln
ln ln ln
ln ln ) ln(
8.1.2 Equazioni di secondo grado
a
ac b b
x c bx ax
2
4
0
2
2
= = + +
8.1.3 Vettori e matrici
Una matrice una rappresentazione rettangolare di un insieme di elementi caratterizzati da
un doppio pedice: il primo rappresenta la riga, il secondo la colonna:
4 ; 2 ; 3 ; 1
4 2
3 1
22 21 12 11
22 21
12 11
= = = =
(
=
(
= a a a a
a a
a a
A
Un vettore una matrice ad una sola riga (o ad una sola colonna):
| | | | 2 ; 1 2 1
2 1 2 1
= = = = x x x x X
Il trasposto di una matrice si ottiene scambiando
ij
a con
ji
a :
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
318
(
=
(
=
(
=
4 3
2 1
4 2
3 1
22 21
12 11 T
A
a a
a a
A
Due matrici possono essere moltiplicate purch il numero delle colonne della prima matrice
sia uguale al numero delle righe della seconda matrice. La matrice risultante ha lo stesso
numero di righe della prima matrice e lo stesso numero di colonne della seconda matrice.
Gli elementi ij della matrice prodotto si ottengono moltiplicando gli elementi della i-esima
riga della prima matrice per gli elementi della j-esima colonna della seconda matrice e
sommando i risultati:
| |
=
= + =
(
=
2
1
2 2 1 1
2
1
2 1
i
i i
b x b x b x
b
b
x x XB
Il prodotto di due vettori rappresenta quindi in forma compatta una regola di miscelazione
lineare.
Nel caso di matrici:
| | | |
22 2 21 1 12 2 11 1
22 21
12 11
2 1
a x a x a x a x
a a
a a
x x XA + + =
(
=
Risulta facile dimostrare che, se
ji ij
a a = :
= =
=
2
1
2
1 i j
ij i j
T
a x x XAX
che rappresenta in forma compatta una regola di miscelazione quadratica.
8.1.4 Derivate, differenziali ed integrali
Nelle relazioni seguenti a e b indicano delle generiche costanti, f(x) e g(x) sono generiche
funzioni della variabile indipendente x, mentre c rappresenta una costante di integrazione.
1
=
n
n
nx
dx
dx
x
x
e
dx
de
=
x dx
x d 1 ln
=
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
319
( ) ( )
dx
x f d
a
dx
x af d ) ( ) (
=
( ) ( ) ( )
dx
x g d
dx
x f d
dx
x g x f d ) ( ) ( ) ( ) (
+ =
+
( ) ( ) ( )
dx
x f d
x g
dx
x g d
x f
dx
x g x f d ) (
) (
) (
) (
) ( ) (
+ =
( ) ( ) ( ) ( )
( )
( )
( ) ( )
( )
( )
( ) ( ) ( )
=
=
=
dx
x g d
x g dx
x g d
dx
x g d
x g
dx
x g d
dx
x g d
x g d
x g f d
dx
x g f d
) (
) (
1 ) ( ln
) (
) ( exp
) ( exp
) (
) (
) ( ) (
( ) dy
y
y x f
dx
x
y x f
y x f d
x
y
|
|
.
|
\
|
+ |
.
|
\
|
=
) , ( ) , (
) , (
Il pedice delle derivate parziali indica che la derivazione viene effettuata mantenendo
costanti le variabili indicate a pedice.
c
n
x
dx x
n
n
+
+
=
+
1
1
c x dx
x
+ =
ln
1
c e
a
dx e
ax ax
+ =
1
c b ax
a
c t
a a
dt
t
dx
b ax
b ax t
+ + = + = =
+
+ =
ln
1
ln
1 1 1
( ) ( )
= dx x g
dx
x f d
x g x f dx
dx
x g d
x f ) (
) (
) ( ) (
) (
) (
8.1.5 Approssimazioni ed interpolazioni
E' spesso utile trovare una funzione g(x) che interpoli o approssimi al meglio una diversa
funzione f(x). Ci essenzialmente dovuto a due motivi: sostituire una f(x) scomoda da
manipolare con una g(x) pi comoda; oppure ricavare il valore approssimato di una f(x)
nota in forma tabulare in un punto non compreso nella tabella.
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
320
8.1.5.1 Espansione in serie di Taylor
In un intorno del punto x, una funzione f(x) pu essere approssimata dalla relazione:
( ) + =
=
=
x x
dx
x df
x f x g x f
x x
x x
) (
) ( ) ( ) (
dove
=x x
dx
x df ) (
la derivata calcolata nel punto x (e quindi un numero, e non una
funzione, cos come
=x x
x f ) ( che il valore della funzione nel punto x) e sono stati
trascurati i termini di ordine superiore al primo
194
. In pratica, con la relazione precedente, si
approssima la f(x) con la sua retta tangente nel punto (x, f(x)), o, in altri termini, si
linearizza landamento della f(x) nellintorno di x.
8.1.5.2 Interpolazione lineare
Data una f(x) nota in forma tabulare per N valori:
n x
n
f(x
n
)
1 x
1
f(x
1
)
2 x
2
f(x
2
)
j-1 x
j-1
f(x
j-1
)
j x
j
f(x
j
)
j+1 x
j+1
f(x
j+1
)
N x
N
f(x
N
)
possibile calcolarne il valore approssimato per un valore di x non compreso in tabella
assumendo che la f(x) vari linearmente tra i valori noti. Quindi, se x contenuto
nell'intervallo [x
j
, x
j+1
]:
) (
) ( ) (
) ( ) ( ) (
1
1
j
j j
j j
j
x x
x x
x f x f
x f x g x f
+ =
+
+
Nel caso di una funzione di due variabili la procedura di interpolazione analoga: prima si
interpola lungo una variabile, e poi lungo la seconda. Data quindi una f(x,y) nota in forma
tabulare per N valori:
194
Cio quei termini che contengono la differenza (x-x) elevata a potenze maggiori di uno. Poich
tale differenza molto piccola (stiamo approssimando la funzione f(x) in un intorno circoscritto di x)
(x-x) >> (x-x)
2
>> (x-x)
3
>>
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
321
y
m
y
1
y
2
y
k-1
y
k
y
k+1
y
M
x
n
x
1
f(x
1
, y
1
) f(x
1
, y
2
) f(x
1
, y
k-1
) f(x
1
, y
k
) f(x
1
, y
k+1
) f(x
1
, y
M
)
x
2
f(x
2
, y
1
) f(x
2
, y
2
) f(x
2
, y
k-1
) f(x
2
, y
k
) f(x
2
, y
k+1
) f(x
2
, y
M
)
x
j-1
f(x
j-1
, y
1
) f(x
j-1
, y
2
) f(x
j-1
, y
k-1
) f(x
j-1
, y
k
) f(x
j-1
, y
k+1
) f(x
j-1
, y
M
)
x
j
f(x
j
, y
1
) f(x
j
, y
2
) f(x
j
, y
k-1
) f(x
j
, y
k-1
) f(x
j
, y
k-1
) f(x
j
, y
M
)
x
j+1
f(x
j+1
, y
1
) f(x
j+1
, y
2
) f(x
j+1
, y
k-1
) f(x
j+1
, y
k
) f(x
j+1
, y
k+1
) f(x
j+1
, y
M
)
x
N
f(x
N
, y
1
) f(x
N
, y
2
) f(x
N
, y
k-1
) f(x
N
, y
k
) f(x
N
, y
k+1
) f(x
N
, y
M
)
possibile calcolarne il valore approssimato per una coppia di valori (x, y) non compresa in
tabella assumendo che la f(x,y) vari linearmente tra i valori noti. Quindi, se x contenuto
nell'intervallo [x
j
, x
j+1
] e y contenuto nell'intervallo [y
k
, y
k+1
] prima si interpola, per
esempio, lungo x per calcolare il valore approssimato di f(x, y
k
) e di f(x, y
k+1
):
) (
) , ( ) , (
) , ( ) , ( ) , (
1
1
j
j j
k j k j
k j k k
x x
x x
y x f y x f
y x f y x g y x f
+ =
+
+
) (
) , ( ) , (
) , ( ) , ( ) , (
1
1 1 1
1 1 1 j
j j
k j k j
k j k k
x x
x x
y x f y x f
y x f y x g y x f
+ =
+
+ + +
+ + +
Questi due nuovi valori della funzione vengono poi interpolati nuovamente (ma questa
volta lungo y) per calcolare il valore desiderato della funzione f(x,y):
) (
) , ( ) , (
) , ( ) , ( ) , (
1
1
k
k k
k k
k
y y
y y
y x f y x f
y x f y x g y x f
+ =
+
+
8.1.5.3 Problematiche connesse col calcolo numerico
Nellambito delle applicazioni e degli esercizi proposti nel testo a volte necessario trovare
la soluzione di problemi formalizzati in equazioni non risolubili analiticamente. L'impiego
di metodi numerici (cio approssimati) per ricercare una soluzione approssimata a tali
problemi non pu, in generale, essere evitato. Sia le moderne calcolatrici sia molti
programmi applicativi disponibili su personal computer forniscono strumenti per la
risoluzione numerica di diversi problemi (quali per esempio la ricerca di radici di equazioni
algebriche o di sistemi di equazioni algebriche). Poich lo studio dei metodi numerici esula
dagli obiettivi di questo testo, non viene riportato alcun dettaglio circa tali metodi
195
, ma
195
Per una trattazione dei metodi numerici si vedano per esempio: K.E. Atkinson "Elementary
Numerical Analysis", J. Wiley & Sons, N.Y. (1985); V. Comincioli "Metodi numerici e statistici per
le scienze applicate", Casa Editrice Ambrosiana, Milano (1992); G. Buzzi Ferraris "Metodi numerici
e Software in C++", Addison-Wesley, N.Y. (1998).
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
322
solo un accenno ai problemi intrinsecamente connessi col calcolo numerico, cio con
lutilizzare dei numeri invece che dei simboli. Tali numeri sono inevitabilmente
approssimati in quanto una calcolatrice o un calcolatore in grado di memorizzare solo un
numero finito di cifre. Gli esempi riportati di seguito illustrano le possibili conseguenze di
tale problema.
Un primo esempio concerne il calcolo degli elementi di una successione con una
calcolatrice tascabile. Si consideri la successione numerica 1 78
1
=
+ n n
S S con valore
iniziale 77 1
0
= S . Tale successione fornisce sempre il valore di 1/77 per tutti i suoi termini.
In realt, se si calcola il valore di ... , ,
2 1
S S partendo dal valore di
0
S dato con una
normale calcolatrice, la successione diverge dopo pochi cicli.
Un secondo esempio riguarda invece la soluzione di un sistema algebrico lineare. In
funzione della procedura di soluzione utilizzata con una calcolatrice tascabile il seguente
sistema:
= +
= +
2 y x
1 y x 10
20
presenta soluzioni diverse, come mostrato nel seguito dove si riportano i risultati ottenuti
eseguendo i calcoli con una calcolatrice tascabile nell'ordine indicato.
1) soluzione errata:
= +
2 y
10
y 1
10
y 1
x
20
20
= +
=
20 20
20
10 2 10 y 1
10
y 1
x
=
=
1
10 1
10 2 1
y
0
10
1 1
x
20
20
20
E' evidente che la soluzione trovata (x = 0; y = 1) non soddisfa il sistema iniziale:
= +
= +
2 1 0
1 1 0 10
20
=
=
2 1
1 1
2) soluzione corretta:
( )
=
= +
y 2 x
1 y y 2 10
20
= =
=
1 1 2 x
1
10 1
10 2 1
y
20
20
Questa seconda soluzione (x = 1; y = 1) invece una buona approssimazione della
soluzione vera.
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
323
8.2 Differenziali esatti e funzioni di stato
La descrizione matematica delle trasformazioni di un sistema termodinamico spesso
conduce ad espressioni differenziali del tipo:
=
= + + +
n
i
i i n n
dx f dx f dx f dx f
1
2 2 1 1
...
dove
i
x sono le variabili indipendenti del sistema termodinamico, mentre
) ,..., , (
2 1 n i
x x x f sono funzioni delle variabili indipendenti.
Quando possibile eguagliare la relazione precedente al differenziale di una nuova
funzione ) ,..., , (
2 1 n
x x x y dipendente dalle stesse variabili indipendenti, allora lespressione
differenziale precedente detta esatta e si pu scrivere:
=
= + + + =
n
i
i i n n
dx f dx f dx f dx f dy
1
2 2 1 1
...
Sfruttando la relazione tra il differenziale di una funzione e le derivate parziali della
funzione stessa:
|
|
.
|
\
|
=
=
|
|
.
|
\
|
+ +
|
|
.
|
\
|
+
|
|
.
|
\
|
=
n
i
i
x
i
n
x x
n
x x x x x
dx
x
y
dx
x
y
dx
x
y
dx
x
y
dy
i j
n n n
1
,...,
2
,..., ,
2
1
,...,
1
1 1 3 1 2
...
Poich le
i
x sono variabili indipendenti, ne consegue che:
1 1 3 1 2
,..., ,..., ,
2
2
,...,
1
1
...; ; ;
|
|
.
|
\
|
=
|
|
.
|
\
|
=
|
|
.
|
\
|
=
n n n
x x
n
n
x x x x x
x
y
f
x
y
f
x
y
f
Ciascuna
i
f e la corrispondente variabile indipendente
i
x sono dette coniugate luna con
laltra.
Se la funzione ) ,..., , (
2 1 n
x x x y e le sue derivate sono continue, allora per ogni coppia di
variabili indipendenti ) ; (
l k
x x vale la relazione:
k l l k
x x
y
x x
y
2 2
Ricordando le relazioni:
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
324
k i l i
l i k i
x
k
l
l k
x
l
l
x
l
k
k l
x
k
k
x
f
x x
y
x
y
f
x
f
x x
y
x
y
f
|
|
.
|
\
|
|
|
.
|
\
|
=
|
|
.
|
\
|
|
|
.
|
\
|
=
2
2
si ricava limportante relazione tra due coppie qualsiasi di variabili coniugate ) ; (
l l
x f e
) ; (
k k
x f :
k i l i
x
k
l
x
l
k
x
f
x
f
|
|
.
|
\
|
=
|
|
.
|
\
|
Se il differenziale di una funzione ) ,..., , (
2 1 n
x x x y esatto allora il valore dellintegrale
=
= =
B
A
n
i
i i
B
A
dx f dy y
1
indipendente dal cammino seguito per passare dallo stato A
allo stato B. Inoltre, lintegrale su di un qualsiasi cammino chiuso (o ciclico)
=
=
n
i
i i
dx f dy
1
nullo. Le funzioni termodinamiche il cui differenziale noto
sperimentalmente essere esatto godono di queste propriet e sono chiamate funzioni di
stato.
8.3 Sistemi con due variabili indipendenti
In questa sezione vengono richiamate alcune relazioni tra derivate parziali per il caso, di
particolare importanza nel campo della termodinamica, di uno stato che pu essere
completamente definito da due variabili di stato (per esempio, y e z) indipendenti. Ne
consegue che in questo caso ogni altra variabile di stato, per esempio x, legata a y e z da
una relazione funzionale del tipo:
0 ) , , ( = z y x f
Poich ciascuna coppia di variabili pu essere arbitrariamente scelta come indipendente,
questa relazione funzionale pu essere posta nelle tre forme alternative:
) , (
) , (
) , (
y x z z
z x y y
z y x x
=
=
=
Considerando arbitrariamente le prime due relazioni devono valere le relazioni generali:
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
325
dz
z
y
dx
x
y
dy
dz
z
x
dy
y
x
dx
x z
y
z
|
.
|
\
|
+ |
.
|
\
|
=
|
.
|
\
|
+
|
|
.
|
\
|
=
Eliminando dy da queste relazioni si ottiene:
0 1 =
(
(
|
.
|
\
|
+ |
.
|
\
|
|
|
.
|
\
|
+
(
(
|
.
|
\
|
|
|
.
|
\
|
dz
z
x
z
y
y
x
dx
x
y
y
x
y x
z
z
z
Poich x e z sono variabili indipendenti, lo sono anche le loro variazioni infinitesime e
quindi necessariamente i due termini tra parentesi quadre devono essere nulli. Ne consegue
una prima importante relazione:
1
|
.
|
\
|
=
|
|
.
|
\
|
z
z
x
y
y
x
che, sostituita nel secondo termine tra parentesi quadra, fornisce una seconda importante
relazione:
x
y
z
y
z
z
x
y
x
|
|
.
|
\
|
|
.
|
\
|
=
|
|
.
|
\
|
Dividendo il differenziale di x per il differenziale di una quarta variabile di stato, w, si
ottiene:
dw
dz
z
x
dw
dy
y
x
dw
dx
y
z
|
.
|
\
|
+
|
|
.
|
\
|
=
Restringendo questa equazione al caso di z = cost si ottiene:
z
z
z
w
y
y
x
w
x
|
.
|
\
|
|
|
.
|
\
|
= |
.
|
\
|
da cui ne consegue unaltra importante relazione:
z
z
z
y
w
w
x
y
x
|
|
.
|
\
|
|
.
|
\
|
=
|
|
.
|
\
|
Se si considera ora ) , ( w y x , il suo differenziale risulta:
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
326
dw
w
x
dy
y
x
dx
y
w
|
.
|
\
|
+
|
|
.
|
\
|
=
Dividendo questa relazione per dy con la restrizione z=costante si ottiene unultima
importante relazione:
z
y
w z
y
w
w
x
y
x
y
x
|
|
.
|
\
|
|
.
|
\
|
+
|
|
.
|
\
|
=
|
|
.
|
\
|
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
327
9 Appendice B
Tabelle termodinamiche dellacqua
Le tabelle seguenti riportano i valori del volume specifico massico, v [cm
3
g
-1
], dellenergia
interna specifica massica, u [kJ kg
-1
], dellentalpia interna specifica massica, h
[kJ kg
-1
] e
dellentropia interna specifica massica, s [kJ K
-1
kg
-1
] del vapor dacqua saturo e
dellacqua liquida satura a diversi valori di temperatura, T [K].
La differenza tra i due valori ovviamente il di evaporazione (per esempio,
] kg [kJ 5 , 2442 8 , 104 3 , 2547 ) [K] 15 , 298 (
) [K] 15 , 298 (
) [K] 15 , 298 (
1
= = =
L V
ev
h h h ).
Per ogni valore di temperatura viene anche riportata la pressione di equilibrio, cio le
tensione di vapore dellacqua a quella temperatura, P [kPa] (per esempio,
[kPa] 166 , 3 ) [K] 15 , 298 (
0
= P ).
T P
L
v
V
v
L
u
V
u
L
h
V
h
L
s
V
s
273,15 0,611 1,000 206300 -0,04 2375,6 -0,04 2501,6 0,0000 9,1578
273,16 0,611 1,000 206200 0,00 2375,6 0,00 2501,6 0,0000 9,1575
274,15 0,657 1,000 192600 4,17 2376,9 4,17 2503,4 0,0153 9,1311
275,15 0,705 1,000 179900 8,39 2378,3 8,39 2505,2 0,0306 9,1047
276,15 0,757 1,000 168200 12,60 2379,7 12,60 2507,1 0,0459 9,0785
277,15 0,813 1,000 157300 16,80 2381,1 16,80 2508,9 0,0611 9,0526
278,15 0,872 1,000 147200 21,01 2382,4 21,01 2510,7 0,0762 9,0269
279,15 0,935 1,000 137800 25,21 2383,8 25,21 2512,6 0,0913 9,0014
280,15 1,001 1,000 129100 29,41 2385,2 29,41 2514,4 0,1063 8,9762
281,15 1,072 1,000 121000 33,60 2386,6 33,60 2516,2 0,1213 8,9513
282,15 1,147 1,000 113400 37,80 2387,9 37,80 2518,1 0,1362 8,9265
283,15 1,227 1,000 106400 41,99 2389,3 41,99 2519,9 0,1510 8,9020
284,15 1,312 1,000 99910 46,18 2390,7 46,19 2521,7 0,1658 8,8776
285,15 1,401 1,000 93840 50,38 2392,1 50,38 2523,6 0,1805 8,8536
286,15 1,497 1,001 88180 54,56 2393,4 54,57 2525,4 0,1952 8,8297
287,15 1,597 1,001 82900 58,75 2394,8 58,75 2527,2 0,2098 8,8060
288,15 1,704 1,001 77980 62,94 2396,2 62,94 2529,1 0,2243 8,7826
289,15 1,817 1,001 73380 67,12 2397,6 67,13 2530,9 0,2388 8,7593
290,15 1,936 1,001 69090 71,31 2398,9 71,31 2532,7 0,2533 8,7363
291,15 2,062 1,001 65090 75,49 2400,3 75,50 2534,5 0,2677 8,7135
292,15 2,196 1,002 61340 79,68 2401,7 79,68 2536,4 0,2820 8,6908
293,15 2,337 1,002 57840 83,86 2403,0 83,86 2538,2 0,2963 8,6684
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
328
T P
L
v
V
v
L
u
V
u
L
h
V
h
L
s
V
s
294,15 2,485 1,002 54560 88,04 2404,4 88,04 2540,0 0,3105 8,6462
295,15 2,642 1,002 51490 92,22 2405,8 92,23 2541,8 0,3247 8,6241
296,15 2,808 1,002 48620 96,40 2407,1 96,41 2543,6 0,3389 8,6023
297,15 2,982 1,003 45930 100,6 2408,5 100,6 2545,5 0,3530 8,5806
298,15 3,166 1,003 43400 104,8 2409,9 104,8 2547,3 0,3670 8,5592
299,15 3,360 1,003 41030 108,9 2411,2 108,9 2549,1 0,3810 8,5379
300,15 3,564 1,003 38810 113,1 2412,6 113,1 2550,9 0,3949 8,5168
301,15 3,778 1,004 36730 117,3 2414,0 117,3 2552,7 0,4088 8,4959
302,15 4,004 1,004 34770 121,5 2415,3 121,5 2554,5 0,4227 8,4751
303,15 4,241 1,004 32930 125,7 2416,7 125,7 2556,4 0,4365 8,4546
304,15 4,491 1,005 31200 129,8 2418,0 129,8 2558,2 0,4503 8,4342
305,15 4,753 1,005 29570 134,0 2419,4 134,0 2560,0 0,4640 8,4140
306,15 5,029 1,005 28040 138,2 2420,8 138,2 2561,8 0,4777 8,3939
307,15 5,318 1,006 26600 142,4 2422,1 142,4 2563,6 0,4913 8,3740
308,15 5,622 1,006 25240 146,6 2423,5 146,6 2565,4 0,5049 8,3543
309,15 5,940 1,006 23970 150,7 2424,8 150,7 2567,2 0,5184 8,3348
310,15 6,274 1,007 22760 154,9 2426,2 154,9 2569,0 0,5319 8,3154
311,15 6,624 1,007 21630 159,1 2427,5 159,1 2570,8 0,5453 8,2962
312,15 6,991 1,007 20560 163,3 2428,9 163,3 2572,6 0,5588 8,2772
313,15 7,375 1,008 19550 167,4 2430,2 167,5 2574,4 0,5721 8,2583
314,15 7,777 1,008 18590 171,6 2431,6 171,6 2576,2 0,5854 8,2395
315,15 8,198 1,009 17690 175,8 2432,9 175,8 2577,9 0,5987 8,2209
316,15 8,639 1,009 16840 180,0 2434,2 180,0 2579,7 0,6120 8,2025
317,15 9,100 1,00 16040 184,2 2435,6 184,2 2581,5 0,6252 8,1842
318,15 9,582 1,010 15280 188,3 2436,9 188,4 2583,3 0,6383 8,1661
319,15 10,09 1,010 14560 192,5 2438,3 192,5 2585,1 0,6514 8,1481
320,15 10,61 1,011 13880 196,7 2439,6 196,7 2586,9 0,6645 8,1302
321,15 11,16 1,011 13230 200,9 2440,9 200,9 2588,6 0,6776 8,1125
322,15 11,74 1,012 12620 205,1 2442,3 205,1 2590,4 0,6906 8,0950
323,15 12,34 1,012 12050 209,2 2443,6 209,3 2592,2 0,7035 8,0776
324,15 12,96 1,013 11500 213,4 2444,9 213,4 2593,9 0,7164 8,0603
325,15 13,61 1,013 10980 217,6 2446,2 217,6 2595,7 0,7293 8,0432
326,15 14,29 1,014 10490 221,8 2447,6 221,8 2597,5 0,7422 8,0262
327,15 15,00 1,014 10020 226,0 2448,9 226,0 2599,2 0,7550 8,0093
328,15 15,74 1,015 9578,9 230,2 2450,2 230,2 2601,0 0,7677 7,9925
329,15 16,51 1,015 9158,7 234,3 2451,5 234,4 2602,7 0,7804 7,9759
330,15 17,31 1,016 8759,8 238,5 2452,8 238,5 2604,5 0,7931 7,9595
331,15 18,15 1,016 8380,8 242,7 2454,1 242,7 2606,2 0,8058 7,9431
332,15 19,02 1,017 8020,8 246,9 2455,4 246,9 2608,0 0,8184 7,9269
333,15 19,92 1,017 7678,5 251,1 2456,8 251,1 2609,7 0,8310 7,9108
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
329
T P
L
v
V
v
L
u
V
u
L
h
V
h
L
s
V
s
334,15 20,86 1,018 7353,2 255,3 2458,1 255,3 2611,4 0,8435 7,8948
335,15 21,84 1,018 7043,7 259,4 2459,4 259,5 2613,2 0,8560 7,8790
336,15 22,86 1,019 6749,3 263,6 2460,7 263,6 2614,9 0,8685 7,8633
337,15 23,91 1,019 6469,0 267,8 2462,0 267,8 2616,6 0,8809 7,8477
338,15 25,01 1,020 6202,3 272,0 2463,2 272,0 2618,4 0,8933 7,8322
339,15 26,15 1,020 5948,2 276,2 2464,5 276,2 2620,1 0,9057 7,8168
340,15 27,33 1,021 5706,2 280,4 2465,8 280,4 2621,8 0,9180 7,8015
341,15 28,56 1,022 5475,6 284,6 2467,1 284,6 2623,5 0,9303 7,7864
342,15 29,84 1,022 5255,8 288,8 2468,4 288,8 2625,2 0,9426 7,7714
343,15 31,16 1,023 5046,3 292,9 2469,7 293,0 2626,9 0,9548 7,7565
344,15 32,53 1,023 4846,4 297,1 2470,9 297,2 2628,6 0,9670 7,7417
345,15 33,96 1,024 4655,7 301,3 2472,2 301,4 2630,3 0,9792 7,7270
346,15 35,43 1,025 4473,7 305,5 2473,5 305,5 2632,0 0,9913 7,7124
347,15 36,96 1,025 4300,0 309,7 2474,8 309,7 2633,7 1,0034 7,6979
348,15 38,55 1,026 4134,1 313,9 2476,0 313,9 2635,4 1,0154 7,6835
349,15 40,19 1,027 3975,7 318,1 2477,3 318,1 2637,1 1,0275 7,6693
350,15 41,89 1,027 3824,3 322,3 2478,5 322,3 2638,7 1,0395 7,6551
351,15 43,65 1,028 3679,6 326,5 2479,8 326,5 2640,4 1,0514 7,6410
352,15 45,47 1,029 3541,3 330,7 2481,1 330,7 2642,1 1,0634 7,6271
353,15 47,36 1,029 3409,1 334,9 2482,3 334,9 2643,8 1,0753 7,6132
354,15 49,31 1,030 3282,6 339,1 2483,5 339,1 2645,4 1,0871 7,5995
355,15 51,33 1,031 3161,6 343,3 2484,8 343,3 2647,1 1,0990 7,5858
356,15 53,42 1,031 3045,8 347,5 2486,0 347,5 2648,7 1,1108 7,5722
357,15 55,57 1,032 2935,0 351,7 2487,3 351,7 2650,4 1,1225 7,5587
358,15 57,80 1,033 2828,8 355,9 2488,5 355,9 2652,0 1,1343 7,5454
359,15 60,11 1,033 2727,2 360,1 2489,7 360,1 2653,6 1,1460 7,5321
360,15 62,49 1,034 2629,8 364,3 2490,9 364,3 2655,3 1,1577 7,5189
361,15 64,95 1,035 2536,5 368,5 2492,2 368,5 2656,9 1,1693 7,5058
362,15 67,49 1,035 2447,0 372,7 2493,4 372,7 2658,5 1,1809 7,4928
363,15 70,11 1,036 2361,3 376,9 2494,6 376,9 2660,1 1,1925 7,4799
364,15 72,81 1,037 2279,1 381,1 2495,8 381,1 2661,7 1,2041 7,4670
365,15 75,61 1,038 2200,2 385,3 2497,0 385,4 2663,4 1,2156 7,4543
366,15 78,49 1,038 2124,5 389,5 2498,2 389,6 2665,0 1,2271 7,4416
367,15 81,46 1,039 2051,9 393,7 2499,4 393,8 2666,6 1,2386 7,4291
368,15 84,53 1,040 1982,2 397,9 2500,6 398,0 2668,1 1,2501 7,4166
369,15 87,69 1,041 1915,3 402,1 2501,8 402,2 2669,7 1,2615 7,4042
370,15 90,94 1,041 1851,0 406,3 2503,0 406,4 2671,3 1,2729 7,3919
371,15 94,30 1,042 1789,3 410,5 2504,1 410,6 2672,9 1,2842 7,3796
372,15 97,76 1,043 1730,0 414,7 2505,3 414,8 2674,4 1,2956 7,3675
373,15 101,33 1,044 1673,0 419,0 2506,5 419,1 2676,0 1,3069 7,3554
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
330
T P
L
v
V
v
L
u
V
u
L
h
V
h
L
s
V
s
375,15 108,78 1,045 1565,5 427,4 2508,8 427,5 2679,1 1,3294 7,3315
377,15 116,68 1,047 1466,2 435,8 2511,1 435,9 2682,2 1,3518 7,3078
379,15 125,04 1,049 1374,2 444,3 2513,4 444,4 2685,3 1,3742 7,2845
381,15 133,90 1,050 1288,9 452,7 2515,7 452,9 2688,3 1,3964 7,2615
383,15 143,27 1,052 1209,9 461,2 2518,0 461,3 2691,3 1,4185 7,2388
385,15 153,16 1,054 1136,6 469,6 2520,2 469,8 2694,3 1,4405 7,2164
387,15 163,62 1,055 1068,5 478,1 2522,4 478,3 2697,2 1,4624 7,1942
389,15 174,65 1,057 1005,2 486,6 2524,6 486,7 2700,2 1,4842 7,1723
391,15 186,28 1,059 946,3 495,0 2526,8 495,2 2703,1 1,5060 7,1507
393,15 198,54 1,061 891,5 503,5 2529,0 503,7 2706,0 1,5276 7,1293
395,15 211,45 1,062 840,5 512,0 2531,1 512,2 2708,8 1,5491 7,1082
397,15 225,04 1,064 792,8 520,5 2533,2 520,7 2711,6 1,5706 7,0873
399,15 239,33 1,066 748,4 529,0 2535,3 529,2 2714,4 1,5919 7,0666
401,15 254,35 1,068 706,9 537,5 2537,4 537,8 2717,2 1,6132 7,0462
403,15 270,13 1,070 668,1 546,0 2539,4 546,3 2719,9 1,6344 7,0261
405,15 286,70 1,072 631,9 554,5 2541,4 554,8 2722,6 1,6555 7,0061
407,15 304,07 1,074 598,0 563,1 2543,4 563,4 2725,3 1,6765 6,9864
409,15 322,29 1,076 566,2 571,6 2545,4 572,0 2727,9 1,6974 6,9669
411,15 341,38 1,078 536,4 580,2 2547,4 580,5 2730,5 1,7182 6,9475
413,15 361,38 1,080 508,5 588,7 2549,3 589,1 2733,1 1,7390 6,9284
415,15 382,31 1,082 482,3 597,3 2551,2 597,7 2735,6 1,7597 6,9095
417,15 404,20 1,084 457,7 605,9 2553,1 606,3 2738,1 1,7803 6,8908
419,15 427,09 1,086 434,6 614,4 2554,9 614,9 2740,6 1,8008 6,8723
421,15 451,01 1,089 412,9 623,0 2556,8 623,5 2743,0 1,8213 6,8539
423,15 476,00 1,091 392,4 631,6 2558,6 632,1 2745,4 1,8416 6,8358
425,15 502,08 1,093 373,2 640,2 2560,3 640,8 2747,7 1,8619 6,8178
427,15 529,29 1,095 355,1 648,9 2562,1 649,4 2750,0 1,8822 6,8000
429,15 557,67 1,098 338,0 657,5 2563,8 658,1 2752,3 1,9023 6,7823
431,15 587,25 1,100 321,9 666,1 2565,5 666,8 2754,5 1,9224 6,7648
433,15 618,06 1,102 306,8 674,8 2567,1 675,5 2756,7 1,9425 6,7475
435,15 650,16 1,105 292,4 683,5 2568,8 684,2 2758,9 1,9624 6,7303
437,15 683,56 1,107 278,9 692,1 2570,4 692,9 2761,0 1,9823 6,7133
439,15 718,31 1,109 266,1 700,8 2571,9 701,6 2763,1 2,0022 6,6964
441,15 754,45 1,112 254,0 709,5 2573,4 710,4 2765,1 2,0219 6,6796
443,15 792,02 1,114 242,6 718,2 2574,9 719,1 2767,1 2,0416 6,6630
445,15 831,06 1,117 231,7 727,0 2576,4 727,9 2769,0 2,0613 6,6465
447,15 871,60 1,120 221,5 735,7 2577,8 736,7 2770,9 2,0809 6,6302
449,15 913,68 1,122 211,7 744,4 2579,3 745,5 2772,7 2,1004 6,6140
451,15 957,36 1,125 202,5 753,2 2580,6 754,3 2774,5 2,1199 6,5979
453,15 1002,7 1,128 193,8 762,0 2581,9 763,1 2776,3 2,1393 6,5819
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
331
T P
L
v
V
v
L
u
V
u
L
h
V
h
L
s
V
s
455,15 1049,6 1,130 185,5 770,8 2583,2 772,0 2778,0 2,1587 6,5660
457,15 1098,3 1,133 177,6 779,6 2584,5 780,8 2779,6 2,1780 6,5503
459,15 1148,8 1,136 170,2 788,4 2585,7 789,7 2781,2 2,1972 6,5346
461,15 1201,0 1,139 163,1 797,2 2586,9 798,6 2782,8 2,2164 6,5191
463,15 1255,1 1,142 156,3 806,1 2588,1 807,5 2784,3 2,2356 6,5036
465,15 1311,1 1,144 149,9 814,9 2589,2 816,5 2785,7 2,2547 6,4883
467,15 1369,0 1,147 143,8 823,8 2590,2 825,4 2787,1 2,2738 6,4730
469,15 1428,9 1,150 138,0 832,7 2591,3 834,4 2788,4 2,2928 6,4578
471,15 1490,9 1,153 132,4 841,6 2592,3 843,4 2789,7 2,3117 6,4428
473,15 1554,9 1,156 127,2 850,6 2593,2 852,4 2790,9 2,3307 6,4278
475,15 1621,0 1,160 122,1 859,5 2594,1 861,4 2792,1 2,3495 6,4128
477,15 1689,3 1,163 117,3 868,5 2595,0 870,5 2793,2 2,3684 6,3980
479,15 1759,8 1,166 112,8 877,5 2595,8 879,5 2794,3 2,3872 6,3832
481,15 1832,6 1,169 108,4 886,5 2596,6 888,6 2795,3 2,4059 6,3686
483,15 1907,7 1,173 104,2 895,5 2597,3 897,7 2796,2 2,4247 6,3539
485,15 1985,2 1,176 100,26 904,5 2598,0 906,9 2797,1 2,4434 6,3394
487,15 2065,1 1,179 96,46 913,6 2598,7 916,0 2797,9 2,4620 6,3249
489,15 2147,5 1,183 92,83 922,7 2599,3 925,2 2798,6 2,4806 6,3104
491,15 2232,4 1,186 89,36 931,8 2599,8 934,4 2799,3 2,4992 6,2960
493,15 2319,8 1,190 86,04 940,9 2600,3 943,7 2799,9 2,5178 6,2817
495,15 2409,9 1,194 82,86 950,1 2600,8 952,9 2800,5 2,5363 6,2674
497,15 2502,7 1,197 79,82 959,2 2601,2 962,2 2800,9 2,5548 6,2532
499,15 2598,2 1,201 76,91 968,4 2601,5 971,5 2801,4 2,5733 6,2390
501,15 2696,5 1,205 74,12 977,6 2601,8 980,9 2801,7 2,5917 6,2249
503,15 2797,6 1,209 71,45 986,9 2602,1 990,3 2802,0 2,6102 6,2107
505,15 2901,6 1,213 68,89 996,2 2602,3 999,7 2802,2 2,6286 6,1967
507,15 3008,6 1,217 66,43 1005,4 2602,4 1009,1 2802,3 2,6470 6,1826
509,15 3118,6 1,221 64,08 1014,8 2602,5 1018,6 2802,3 2,6653 6,1686
511,15 3231,7 1,225 61,82 1024,1 2602,5 1028,1 2802,3 2,6837 6,1546
513,15 3347,8 1,229 59,65 1033,5 2602,5 1037,6 2802,2 2,7020 6,1406
515,15 3467,2 1,233 57,57 1042,9 2602,4 1047,2 2802,0 2,7203 6,1266
517,15 3589,8 1,238 55,58 1052,3 2602,2 1056,8 2801,8 2,7386 6,1127
519,15 3715,7 1,242 53,66 1061,8 2602,0 1066,4 2801,4 2,7569 6,0987
521,15 3844,9 1,247 51,81 1071,3 2601,8 1076,1 2801,0 2,7752 6,0848
523,15 3977,6 1,251 50,04 1080,8 2601,4 1085,8 2800,4 2,7935 6,0708
525,15 4113,7 1,256 48,33 1090,4 2601,0 1095,5 2799,8 2,8118 6,0569
527,15 4253,4 1,261 46,69 1100,0 2600,5 1105,3 2799,1 2,8300 6,0429
529,15 4396,7 1,266 45,11 1109,6 2600,0 1115,2 2798,3 2,8483 6,0290
531,15 4543,7 1,271 43,60 1119,3 2599,3 1125,0 2797,4 2,8666 6,0150
533,15 4694,3 1,276 42,13 1129,0 2598,6 1134,9 2796,4 2,8848 6,0010
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
332
T P
L
v
V
v
L
u
V
u
L
h
V
h
L
s
V
s
535,15 4848,8 1,281 40,73 1138,7 2597,8 1144,9 2795,3 2,9031 5,9869
537,15 5007,1 1,286 39,37 1148,5 2597,0 1154,9 2794,1 2,9214 5,9729
539,15 5169,3 1,291 38,06 1158,3 2596,1 1165,0 2792,8 2,9397 5,9588
541,15 5335,5 1,297 36,80 1168,2 2595,0 1175,1 2791,4 2,9580 5,9446
543,15 5505,8 1,303 35,59 1178,1 2593,9 1185,2 2789,9 2,9763 5,9304
545,15 5680,2 1,308 34,42 1188,0 2592,7 1105,4 2788,2 2,9947 5,9162
547,15 5858,7 1,314 33,29 1198,0 2591,4 1205,7 2786,5 3,0131 5,9019
549,15 6041,5 1,320 32,20 1208,0 2590,1 1216,0 2784,6 3,0314 5,8876
551,15 6228,7 1,326 31,14 1218,1 2588,6 1226,4 2782,6 3,0499 5,8731
553,15 6420,2 1,332 30,13 1228,3 2587,0 1236,8 2780,4 3,0683 5,8586
555,15 6616,1 1,339 29,14 1238,5 2585,3 1247,3 2778,1 3,0868 5,8440
557,15 6816,6 1,345 28,20 1248,7 2583,5 1257,9 2775,7 3,1053 5,8294
559,15 7021,8 1,352 27,28 1259,0 2581,6 1268,5 2773,2 3,1238 5,8146
561,15 7231,5 1,359 26,39 1269,4 2579,6 1279,2 2770,5 3,1424 5,7997
563,15 7446,1 1,366 25,54 1279,8 2577,5 1290,0 2767,6 3,1611 5,7848
565,15 7665,4 1,373 24,71 1290,3 2575,3 1300,9 2164,6 3,1798 5,7697
567,15 7889,7 1,381 23,90 1300,9 2572,9 1311,8 2761,5 3,1985 5,7545
569,15 8118,9 1,388 23,13 1311,5 2570,4 1322,8 2758,2 3,2173 5,7392
571,15 8353,2 1,396 22,38 1322,2 2567,8 1333,9 2754,7 3,2362 5,7237
573,15 8592,7 1,404 21,65 1333,0 2565,0 1345,1 2751,0 3,2552 5,7081
575,15 8837,4 1,412 20,94 1343,8 2562,1 1356,3 2747,2 3,2742 5,6924
577,15 9087,3 1,421 20,26 1354,8 2559,1 1367,7 2743,2 3,2933 5,6765
579,15 9342,7 1,430 19,60 1365,8 2555,9 1379,1 2739,0 3,3125 5,6604
581,15 9603,6 1,439 18,96 1376,9 2552,5 1390,7 2734,6 3,3318 5,6442
583,15 9870,0 1,448 18,33 1388,1 2549,1 1402,4 2730,0 3,3512 5,6278
585,15 10142,1 1,458 17,73 1399,4 2545,4 1414,2 2725,2 3,3707 5,6111
587,15 10420,0 1,468 17,14 1410,8 2541,6 1426,1 2720,2 3,3903 5,5943
589,15 10703, 1,478 16,57 1422,3 2537,5 1438,1 2714,9 3,4101 5,5772
591,15 10993,4 1,488 16,02 1433,9 2533,3 1450,3 2709,4 3,4300 5,5599
593,15 11289,1 1,500 15,48 1445,7 2528,9 1462,6 2703,7 3,4500 5,5423
595,15 11591,0 1,511 14,96 1457,5 2524,3 1475,1 2697,6 3,4702 5,5244
597,15 11899,2 1,523 14,45 1469,5 2519,4 1487,7 2691,3 3,4906 5,5062
599,15 12213,7 1,535 13,95 1481,7 2514,3 1500,4 2684,6 3,5111 5,4876
601,15 12534,8 1,548 13,46 1494,0 2508,8 1513,4 2677,6 3,5319 5,4685
603,15 12862,5 1,561 12,99 1506,4 2503,1 1526,5 2670,2 3,5528 5,4490
605,15 13197,0 1,575 12,53 1519,1 2497,0 1539,9 2662,3 3,5740 5,4290
607,15 13538,3 1,590 12,08 1531,9 2490,6 1553,4 2654,1 3,5955 5,4084
609,15 13886,7 1,606 11,63 1544,9 2483,7 1567,2 2645,3 3,6172 5,3872
611,15 14242,3 1,622 11,20 1558,1 2476,4 1581,2 2636,0 3,6392 5,3653
613,15 14605,2 1,639 10,78 1571,5 2468,7 1595,5 2626,2 3,6616 5,3427
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
333
T P
L
v
V
v
L
u
V
u
L
h
V
h
L
s
V
s
615,15 14975,5 1,657 10,37 1585,2 2460,5 1610,0 2615,7 3,6844 5,3194
617,15 15353,5 1,676 9,962 1599,2 2451,7 1624,9 2604,7 3,7075 5,2952
619,15 15739,3 1,696 9,566 1613,5 2442,4 1640,2 2593,0 3,7311 5,2702
621,15 16133,1 1,718 9,178 1628,1 2432,6 1655,8 2580,7 3,7553 5,2444
623,15 16535,1 1,741 8,799 1643,0 2422,2 1671,8 2567,7 3,7801 5,2177
625,15 16945,5 1,766 8,420 1659,4 2410,8 1689,3 2553,5 3,8071 5,1893
627,15 17364,4 1,794 8,045 1676,3 2398,7 1707,5 2538,4 3,8349 5,1596
629,15 17792,2 1,824 7,674 1693,4 2385,6 1725,9 2522,1 3,8629 5,1283
631,15 18229,0 1,858 7,306 1710,8 2371,4 1744,7 2504,6 3,8915 5,0953
633,15 18675,1 1,896 6,940 1728,8 2355,8 1764,2 2485,4 3,9210 5,0600
634,15 18901,7 1,917 6,757 1738,0 2347,5 1774,2 2475,2 3,9362 5,0414
635,15 19130,7 1,939 6,573 1747,5 2338,7 1784,6 2464,4 3,9518 5,0220
636,15 19362,1 1,963 6,388 1757,3 2329,3 1795,3 2453,0 3,9679 5,0017
637,15 19596,1 1,988 6,201 1767,4 2319,4 1806,4 2440,9 3,9846 4,9804
638,15 19832,6 2,016 6,012 1778,0 2308,8 1818,0 2428,0 4,0021 4,9579
639,15 20071,6 2,046 5,819 1789,1 2297,3 1830,2 2414,1 4,0205 4,9339
640,15 20313,2 2,080 5,621 1801,0 2284,8 1843,2 2399,0 4,0401 4,9081
641,15 20557,5 2,118 5,416 1813,8 2271,1 1857,3 2382,4 4,0613 4,8801
642,15 20804,4 2,162 5,201 1827,8 2255,7 1872,8 2363,9 4,0846 4,8492
643,15 21054,0 2,214 4,973 1843,6 2238,1 1890,2 2342,8 4,1108 4,8144
644,15 21306,4 2,278 4,723 1862,0 2217,3 1910,5 2317,9 4,1414 4,7738
645,15 21561,6 2,364 4,439 1884,6 2191,2 1935,6 2287,0 4,1794 4,7240
646,15 21819,7 2,496 4,084 1916,0 2154,9 1970,5 2244,0 4,2325 4,6559
647,15 22080,5 2,843 3,466 1983,9 2079,7 2046,7 2156,2 4,3493 4,5185
647,30 22120,0 3,170 3,170 2037,3 2037,3 2107,4 2107,4 4,4429 4,4429
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
335
10 Appendice C
Tabelle di Lee - Kesler
Le tabelle seguenti riportano i valori suggeriti fa Lee e Kesler per il calcolo di: fattore di
compressibilit, Z, entalpia residua adimensionale, ( )
C
R
RT h , entropia residua
adimensionale, R s
R
, coefficiente di fugacit, (che correlato alla energia libera di
Gibbs residua adimensionale dalla relazione ( ) RT g
R
= ln ) con lequazione degli stati
corrispondenti in funzione della temperatura ridotta, T
R
, e della pressione ridotta, P
R
. Larea
pi scura indica la regione di esistenza della fase liquida.
Il calcolo delle diverse funzioni viene effettuato con le relazioni:
1 0
Z Z Z + =
( ) ( ) | | ( ) | |
1 0
C
R
C
R
C
R
RT h RT h RT h + =
| | | |
1 0
R s R s R s
R R R
+ =
( )
1 0
ln ln ln + = = RT g
R
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
336
FATTORE DI COMPRESSIBILIT:
0
Z
P
R
0,0100 0,0500 0,1000 0,2000 0,4000 0,6000 0,8000 1,0000
T
R
0,30 0,0029 0,0145 0,0290 0,0579 0,1158 0,1737 0,2315 0,2892
0,35 0,0026 0,0130 0,0261 0,0522 0,1043 0,1564 0,2084 0,2604
0,40 0,0024 0,0119 0,0239 0,0477 0,0953 0,1429 0,1904 0,2379
0,45 0,0022 0,0110 0,0221 0,0442 0,0882 0,1322 0,1762 0,2200
0,50 0,0021 0,0103 0,0207 0,0413 0,0825 0,1236 0,1647 0,2056
0,55 0,9804 0,0098 0,0195 0,0390 0,0778 0,1166 0,1553 0,1939
0,60 0,9849 0,0093 0,0186 0,0371 0,0741 0,1109 0,1476 0,1842
0,65 0,9881 0,9377 0,0178 0,0356 0,0710 0,1063 0,1415 0,1765
0,70 0,9904 0,9504 0,8958 0,0344 0,0687 0,1027 0,1366 0,1703
0,75 0,9922 0,9598 0,9165 0,0336 0,0670 0,1001 0,1330 0,1656
0,80 0,9935 0,9669 0,9319 0,8539 0,0661 0,0985 0,1307 0,1626
0,85 0,9946 0,9725 0,9436 0,8810 0,0661 0,0983 0,1301 0,1614
0,90 0,9954 0,9768 0,9528 0,9015 0,7800 0,1006 0,1321 0,1630
0,93 0,9959 0,9790 0,9573 0,9115 0,8059 0,6635 0,1359 0,1664
0,95 0,9961 0,9803 0,9600 0,9174 0,8206 0,6967 0,1410 0,1705
0,97 0,9963 0,9815 0,9625 0,9227 0,8338 0,7240 0,5580 0,1779
0,98 0,9965 0,9821 0,9637 0,9253 0,8398 0,7360 0,5887 0,1844
0,99 0,9966 0,9826 0,9648 0,9277 0,8455 0,7471 0,6138 0,1959
1,00 0,9967 0,9832 0,9659 0,9300 0,8509 0,7574 0,6355 0,2901
1,01 0,9968 0,9837 0,9669 0,9322 0,8561 0,7671 0,6542 0,4648
1,02 0,9969 0,9842 0,9679 0,9343 0,8610 0,7761 0,6710 0,5146
1,05 0,9971 0,9855 0,9707 0,9401 0,8743 0,8002 0,7130 0,6026
1,10 0,9975 0,9874 0,9747 0,9485 0,8930 0,8323 0,7649 0,6880
1,15 0,9978 0,9891 0,9780 0,9554 0,9081 0,8576 0,8032 0,7443
1,20 0,9981 0,9904 0,9808 0,9611 0,9205 0,8779 0,8330 0,7858
1,30 0,9985 0,9926 0,9852 0,9702 0,9396 0,9083 0,9764 0,8438
1,40 0,9988 0,9942 0,9884 0,9768 0,9534 0,9298 0,9062 0,8827
1,50 0,9991 0,9954 0,9909 0,9818 0,9636 0,9456 0,9278 0,9103
1,60 0,9993 0,9964 0,9928 0,9856 0,9714 0,9575 0,9439 0,9308
1,70 0,9994 0,9971 0,9943 0,9886 0,9775 0,9667 0,9563 0,9463
1,80 0,9995 0,9977 0,9955 0,9910 0,9823 0,9739 0,9659 0,9583
1,90 0,9996 0,9982 0,9964 0,9929 0,9861 0,9796 0,9735 0,9678
2,00 0,9997 0,9986 0,9972 0,9944 0,9892 0,9842 0,9796 0,9754
2,20 0,9998 0,9992 0,9983 0,9967 0,9937 0,9910 0,9886 0,9865
2,40 0,9990 0,9996 0,9991 0,9983 0,9969 0,9957 0,9948 0,9941
2,60 1,0000 0,9998 0,9997 0,9994 0,9991 0,9990 0,9990 0,9993
2,80 1,0000 1,0000 1,0001 1,0002 1,0007 1,0013 1,0021 1,0031
3,00 1,0000 1,0002 1,0004 1,0008 1,0018 1,0030 1,0043 1,0057
3,50 1,0001 1,0004 1,0008 1,0017 1,0035 1,0055 1,0075 1,0097
4,00 1,0001 1,0005 1,0010 1,0021 1,0043 1,0066 1,0090 1,0115
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
337
P
R
1,0000 1,2000 1,5000 2,0000 3,0000 5,0000 7,0000 10,000
T
R
0,30 0,2892 0,3479 0,4335 0,5775 0,8648 1,4366 2,0048 2,8507
0,35 0,2604 0,3123 0,3901 0,5195 0,7775 1,2902 1,7987 2,5539
0,40 0,2379 0,2853 0,3563 0,4744 0,7095 1,1758 1,6373 2,3211
0,45 0,2200 0,2638 0,3294 0,4384 0,6551 1,0841 1,5077 2,1338
0,50 0,2056 0,2465 0,3077 0,4092 0,6110 1,0094 1,4017 1,9801
0,55 0,1939 0,2323 0,2899 0,3853 0,5747 0,9475 1,3137 1,8520
0,60 0,1842 0,2207 0,2753 0,3657 0,5446 0,8959 1,2398 1,7440
0,65 0,1765 0,2113 0,2634 0,3495 0,5197 0,8526 1,1773 1,6519
0,70 0,1703 0,2038 0,2538 0,3364 0,4991 0,8161 1,1341 1,5729
0,75 0,1656 0,1981 0,2464 0,3260 0,4823 0,7854 1,0787 1,5047
0,80 0,1626 0,1942 0,2411 0,3182 0,4690 0,7598 1,0400 1,4456
0,85 0,1614 0,1924 0,2382 0,3132 0,4591 0,7388 1,0071 1,3943
0,90 0,1630 0,1935 0,2383 0,3114 0,4527 0,7220 0,9793 1,3496
0,93 0,1664 0,1963 0,2405 0,3122 0,4507 0,7138 0,9648 1,3257
0,95 0,1705 0,1998 0,2432 0,3138 0,4501 0,7092 0,9561 1,3108
0,97 0,1779 0,2055 0,2474 0,3164 0,4504 0,7052 0,9480 1,2968
0,98 0,1844 0,2097 0,2503 0,3182 0,4508 0,7035 0,9442 1,2901
0,99 0,1959 0,2154 0,2538 0,3204 0,4514 0,7018 0,9406 1,2835
1,00 0,2901 0,2237 0,2583 0,3229 0,4522 0,7004 0,9372 1,2772
1,01 0,4648 0,2370 0,2640 0,326,0 0,4533 0,6991 0,9339 1,2710
1,02 0,5146 0,2629 0,2715 0,3297 0,4547 0,6980 0,9307 1,2650
1,05 0,6026 0,4437 0,3131 0,3452 0,4604 0,6956 0,9222 1,2481
1,10 0,6880 0,5984 0,4580 0,3953 0,4770 0,6950 0,9110 1,2232
1,15 0,7443 0,6803 0,5798 0,4760 0,5042 0,6987 0,9033 1,2021
1,20 0,7858 0,7363 0,6605 0,5605 0,5425 0,7069 0,8990 1,1844
1,30 0,8438 0,8111 0,7624 0,6908 0,6344 0,7358 0,8998 1,1580
1,40 0,8827 0,8595 0,8256 0,7753 0,7202 0,7761 0,9112 1,1419
1,50 0,9103 0,8933 0,8689 0,8328 0,7887 0,8200 0,9297 1,1339
1,60 0,9308 0,9180 0,9000 0,8738 0,8410 0,8617 0,9518 1,1320
1,70 0,9463 0,9367 0,9234 0,9043 0,8809 0,8984 0,9745 1,1343
1,80 0,9583 0,9511 0,9413 0,9275 0,9118 0,9297 0,9061 1,1391
1,90 0,9678 0,9624 0,9552 0,9456 0,9359 0,9557 1,0157 1,1452
2,00 0,9754 0,9715 0,9664 0,9599 0,9550 0,9772 1,0328 1,1516
2,20 0,9856 0,9847 0,9826 0,9806 0,9827 1,0094 1,0600 1,1635
2,40 0,9941 0,9936 0,9935 0,9945 1,0011 1,0313 1,0793 1,1728
2,60 0,9993 0,9998 1,0010 1,0040 1,0137 1,0463 1,0926 1,1792
2,80 1,0031 1,0042 1,0063 1,0106 1,0223 1,0565 1,1016 1,1830
3,00 1,0057 1,0074 1,0101 1,0153 1,0284 1,0635 1,1075 1,1848
3,50 1,0097 1,0120 1,0156 1,0221 1,0368 1,0723 1,1138 1,1834
4,00 1,0115 1,0140 1,0179 1,0249 1,0401 1,0747 1,1136 1,1773
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
338
FATTORE DI COMPRESSIBILIT:
1
Z
P
R
0,0100 0,0500 0,1000 0,2000 0,4000 0,6000 0,8000 1,0000
T
R
0,30 -0,0008 -0,0040 -0,0081 -0,0161 -0,0323 -0,0484 -0,0645 -0,0806
0,35 -0,0009 -0,0046 -0,0093 -0,0185 -0,0370 -0,0554 -0,0738 -0,0921
0,40 -0,0010 -0,0048 -0,0095 -0,0190 -0,0380 -0,0570 -0,0758 -0,0946
0,45 -0,0009 -0,0047 -0,0094 -0,0187 -0,0374 -0,0560 -0,0745 -0,0929
0,50 -0,0009 -0,0045 -0,0090 -0,0181 -0,0360 -0,0539 -0,0716 -0,0893
0,55 -0,0314 -0,0043 -0,0086 -0,0172 -0,0343 -0,0513 -0,0682 -0,0849
0,60 -0,0205 -0,0041 -0,0082 -0,0164 -0,0326 -0,0487 -0,0646 -0,0803
0,65 -0,0137 -0,0772 -0,0078 -0,0156 -0,0309 -0,0461 -0,0611 -0,0759
0,70 -0,0093 -0,0507 -0,1161 -0,0148 -0,0294 -0,0438 -0,0579 -0,0718
0,75 -0,0064 -0,0339 -0,0744 -0,0143 -0,0282 -0,0417 -0,0550 -0,0681
0,80 -0,0044 -0,0228 -0,0487 -0,1160 -0,0272 -0,0401 -0,0526 -0,0648
0,85 -0,0029 -0,0152 -0,0319 -0,0715 -0,0268 -0,0391 -0,0509 -0,0622
0,90 -0,0019 -0,0099 -0,0205 -0,0442 -0,1118 -0,0396 -0,0503 -0,0604
0,93 -0,0015 -0,0075 -0,0154 -0,0326 -0,0763 -0,1662 -0,0514 -0,0602
0,95 -0,0012 -0,0062 -0,0126 -0,0262 -0,0589 -0,1110 -0,0540 -0,0607
0,97 -0,0010 -0,0050 -0,0101 -0,0208 -0,0450 -0,0770 -0,1647 -0,0623
0,98 -0,0009 -0,0044 -0,0090 -0,0184 -0,0390 -0,0641 -0,1100 -0,0641
0,99 -0,0008 -0,0039 -0,0079 -0,0161 -0,0335 -0,0531 -0,0796 -0,0680
1,00 -0,0007 -0,0034 -0,0069 -0,0140 -0,0285 -0,0435 -0,0588 -0,0879
1,01 -0,0006 -0,0030 -0,0060 -0,0120 -0,0240 -0,0351 -0,0429 -0,0223
1,02 -0,0005 -0,0026 -0,0051 -0,0102 -0,0198 -0,0277 -0,0303 -0,0062
1,05 -0,0003 -0,0015 -0,0029 -0,0054 -0,0092 -0,0097 -0,0032 0,0220
1,10 0,0000 0,0000 0,0001 0,0007 0,0038 0,0106 0,0236 0,0476
1,15 0,0002 0,0011 0,0023 0,0052 0,0127 0,0237 0,0396 0,0625
1,20 0,0004 0,0019 0,0039 0,0084 0,0190 0,0326 0,0499 0,0719
1,30 0,0006 0,0030 0,0061 0,0125 0,0267 0,0429 0,0612 0,0819
1,40 0,0007 0,0036 0,0072 0,0147 0,0306 0,0477 0,0661 0,0857
1,50 0,0008 0,0039 0,0078 0,0158 0,0323 0,0497 0,0677 0,0864
1,60 0,0008 0,0040 0,0080 0,0162 0,0330 0,0501 0,0677 0,0855
1,70 0,0008 0,0040 0,0081 0,0163 0,0329 0,0497 0,0667 0,0838
1,80 0,0008 0,0040 0,0081 0,0162 0,0325 0,0488 0,0652 0,0814
1,90 0,0008 0,0040 0,0079 0,0159 0,0318 0,0477 0,0635 0,0792
2,00 0,0008 0,0039 0,0078 0,0155 0,0310 0,0464 0,0617 0,0767
2,20 0,0007 0,0037 0,0074 0,0147 0,0293 0,0437 0,0579 0,0719
2,40 0,0007 0,0035 0,0070 0,0139 0,0276 0,0411 0,0544 0,0675
2,60 0,0007 0,0033 0,0066 0,0131 0,0260 0,0387 0,0512 0,0634
2,80 0,0006 0,0031 0,0062 0,0124 0,0245 0,0365 0,0483 0,0598
3,00 0,0006 0,0029 0,0059 0,0117 0,0232 0,0345 0,0456 0,0565
3,50 0,0005 0,0026 0,0052 0,0103 0,0204 0,0303 0,0401 0,0497
4,00 0,0005 0,0023 0,0046 0,0091 0,0182 0,0270 0,0357 0,0443
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
339
P
R
1,0000 1,2000 1,5000 2,0000 3,0000 5,0000 7,0000 10,000
T
R
0,30 -0,0806 -0,0966 -0,1207 -0,1608 -0,2407 -0,3996 -0,5572 -0,7915
0,35 -0,0921 -0,1105 -0,1379 -0,1834 -0,2738 -0,4523 -0,6279 -0,8863
0,40 -0,0946 -0,1134 -0,1414 -0,1879 -0,2799 -0,4603 -0,6365 -0,8936
0,45 -0,0929 -0,1113 -0,1387 -0,1840 -0,2734 -0,4475 -0,6162 -0,8608
0,50 -0,0893 -0,1069 -0,1330 -0,1762 -0,2611 -0,4253 -0,5831 -0,8099
0,55 -0,0849 -0,1015 -0,1263 -0,1669 -0,2465 -0,3991 -0,5446 -0,7521
0,60 -0,0803 -0,0960 -0,1192 -0,1572 -0,2312 -0,3718 -0,5047 -0,6928
0,65 -0,0759 -0,0906 -0,1122 -0,1476 -0,2160 -0,3447 -0,4653 -0,6346
0,70 -0,0118 -0,0855 -0,1057 -0,1385 -0,2013 -0,3184 -0,4270 -0,5785
0,75 -0,0681 -0,0808 -0,0996 -0,1298 -0,1872 -0,2929 -0,3901 -0,5250
0,80 -0,0648 -0,0767 -0,0940 -0,1217 0,1736 -0,2682 -0,3545 -0,4740
0,85 -0,0622 -0,0731 -0,0888 -0,1138 -0,1602 -0,2439 -0,3201 -0,4254
0,90 -0,0604 -0,0701 -0,0840 -0,1059 -0,1463 -0,2195 -0,2862 -0,3788
0,93 -0,0602 -0,0687 -0,0810 -0,1007 -0,1374 -0,2045 -0,2661 -0,3516
0,95 -0,0607 -0,0678 -0,0788 -0,0967 -0,1310 -0,1943 -0,2526 -0,3339
0,97 -0,0623 -0,0669 -0,0759 -0,0921 -0,1240 -0,1837 -0,2391 -0,3163
0,98 -0,0641 -0,0661 -0,0740 -0,0893 -0,1202 -0,1783 -0,2322 -0,3075
0,99 -0,0680 -0,0646 -0,0715 -0,0861 -0,1162 -0,1729 -0,2254 -0,2989
1,00 -0,0879 -0,0609 -0,0678 -0,0824 -0,1118 -0,1672 -0,2185 -0,2902
1,01 -0,0223 -0,0473 -0,0621 -0,0778 -0,1072 -0,1615 -0,2116 -0,2816
1,02 -0,0062 -0,0227 -0,0524 -0,0722 -0,1021 -0,1556 -0,2047 -0,2731
1,05 0,0220 0,1059 0,0451 -0,0432 -0,0838 -0,,370 -0,1835 -0,2476
1,10 0,0476 0,0897 0,1630 0,0698 -0,0373 -0,1021 -0,1469 -0,2056
1,15 0,0625 0,0943 0,1548 0,1667 0,0332 -0,0611 -0,1084 -0,1642
1,20 0,0719 0,0991 0,1477 0,1990 0,1095 -0,0141 -0,0678 -0,1231
1,30 0,0819 0,1048 0,1429 0,1991 0,2079 0,0875 0,0176 -0,0423
1,40 0,0857 0,1063 0,1383 0,1894 0,2397 0,1737 0,1008 0,0350
1,50 0,0854 0,1055 0,1345 0,1806 0,2433 0,2309 0,1717 0,1058
1,60 0,0855 0,1035 0,1303 0,1729 0,2381 0,2631 0,2255 0,1673
1,70 0,0838 0,1008 0,1259 0, 1658 0,2305 0,2788 0,2628 0,2179
1,80 0,0816 0,0978 0,1216 0,1593 0,2224 0,2846 0,2871 0,2576
1,90 0,0792 0,0947 0,1173 0,1532 0,2144 0,2848 0,3017 0,2876
2,00 0,0767 0,0916 0,1133 0, 1476 0,2069 0,2819 0,3097 0,3069
2,20 0,0719 0,0857 0,1057 0,1374 0,1932 0,2720 0,3135 0,3355
2,40 0,0675 0,0803 0,0989 0,1285 0,1812 0,2622 0,3089 0,3459
2,60 0,0634 0,0754 0,0929 0,1207 0,1706 0,2484 0,3009 0,3475
2,80 0,0598 0,0711 0,0876 0,1138 0,1613 0,2372 0,2915 0,3443
3,00 0,0535 0,0672 0,0828 0,1076 0,1529 0,2268 0,2817 0,3385
3,50 0,0497 0,0591 0,0728 0,0949 0,1356 0,2042 0,2584 0,3194
4,00 0,0443 0,0527 0,0651 0,0849 0,1219 0,1857 0,2378 0,2994
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
340
ENTALPIA RESIDUA: [h
R
/(RT
C
)]
0
P
R
0,0100 0,0500 0,1000 0,2000 0,4000 0,6000 0,8000 1,0000
T
R
0,30 -6,045 -6,043 -6,040 -6,034 -6,022 -6,011 -5,999 -5,987
0,35 -5,906 -5,904 -5,901 -5,895 -5,882 -5,870 -5,858 -5,845
0,40 -5,763 -5,761 -5,757 -5,751 -5,738 -5,726 -5,113 -5,700
0,45 -5,615 -5,612 -5,609 -5,603 -5,590 -5,577 -5,564 -5,551
0,50 -5,465 -5,463 -5,459 -5,453 -5,440 -5,427 -5,414 -5,401
0,55 -0,032 -5,312 -5,309 -5,303 -5,290 -5,278 -5,265 -5,252
0,60 -0,027 -5,162 -5,159 -5,153 -5,141 -5,129 -5,116 -5,104
0,65 -0,023 -0,118 -5,008 -5,002 -4,991 -4,980 -4,968 -4,956
0,70 -0,020 -0,101 -0,213 -4,848 -4,838 -4,828 -4,818 -4,808
0,75 -0,017 -0,088 -0,183 -4,687 -4,679 -4,672 -4,664 -4,655
0,80 -0,015 -0,078 -0,160 -0,345 -4,507 -4,504 -4,499 -4,494
0,85 -0,014 -0,069 -0,141 -0,300 -4,309 -4,313 -4,316 -4,316
0,90 -0,012 -0,062 -0,126 -0,264 -0,596 -4,074 -4,094 -4,108
0,93 -0,011 -0,058 -0,118 -0,246 -0,545 -0,960 -3,920 -3,953
0,95 -0,011 -0,056 -0,113 -0,235 -0,516 -0,885 -3,763 -3,825
0,97 -0,011 -0,054 -0,109 -0,225 -0,490 -0,824 -1,356 -3,658
0,98 -0,010 -0,053 -0,107 -0,221 -0,478 -0,797 -1,273 -3,544
0,99 -0,010 -0,052 -0,105 -0,216 -0,466 -0,773 -1,206 -3,376
1,00 -0,010 -0,051 -0,103 -0,212 -0,455 -0,750 -1,151 -2,584
1,01 -0,010 -0,050 -0,101 -0,208 -0,445 -0,721 -1,102 -1,796
1,02 -0,010 -0,049 -0,099 -0,203 -0,434 -0,708 -1,060 -1,627
1,05 -0,009 -0,046 -0,094 -0,192 -0,407 -0,654 -0,955 -1,359
1,10 -0,008 -0,042 -0,086 -0,175 -0,367 -0,581 -0,827 -1,120
1,15 -0,008 -0,039 -0,079 -0,160 -0,334 -0,523 -0,732 -0,968
1,20 -0,007 -0,036 -0,073 -0,148 -0,305 -0,474 -0,657 -0,857
1,30 -0,006 -10,031 -0,063 -0,127 -0,259 -0,399 -0,545 -0,698
1,40 -0,005 -0,027 -0,055 -0,110 -0,224 -0,341 -0,463 -0,588
1,50 -0,005 -0,024 -0,048 -0,097 -0,196 -0,297 -0,400 -0,505
1,60 -0,004 -0,021 -0,043 -0,086 -0,173 -0,261 -0,350 -0,440
1,70 -0,004 -0,019 -0,038 -0,076 -0,153 -0,231 -0,309 -0,387
1,80 -0,003 -0,017 -0,034 -0,068 -0,137 -0,206 -0,275 -0,344
1,90 -0,003 -0,015 -0,031 -0,062 -0,123 -0,185 -0,246 -0,307
2,00 -0,003 -0,014 -0,028 -0,056 -0,111 -0,167 -0,222 -0,276
2,20 -0,002 -0,012 -0,023 -0,046 -0,092 -0,137 -0,182 -0,226
2,40 -0,002 -0,010 -0,019 -0,038 -0,076 -0,114 -0,150 -0,187
2,60 -0,002 -0,008 -0,016 -0,032 -0,064 -0,095 -0,125 -0,155
2,80 -0,001 -0,007 -0,014 -0,027 -0,054 -0,080 -0,105 -0,130
3,00 -0,001 -0,006 -0,011 -0,023 -0,045 -0,067 -0,088 -0,109
3,50 -0,001 -0,004 -0,007 -0,015 -0,029 -0,043 -0,056 -0,069
4,00 -0,000 -0,002 -0,005 -0,009 -0,017 -0,026 -0,033 -0,041
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
341
P
R
1,0000 1,2000 1,5000 2,0000 3,0000 5,0000 7,0000 10,000
T
R
0,30 -5,987 -5,975 -5,957 -5,-927 -5,868 -5,748 -5,628 -5,446
0,35 -5,845 -5,833 -5,814 -5,783 -5,721 -5,595 -5,469 -5,278
0,40 -5,700 -5,687 -5,668 -5,636 -5,572 -5,442 -5,311 -5,113
0,45 -5,551 -5,538 -5,519 -5,486 -5,421 -5,288 -5,154 -5,950
0,50 -5,401 -5,388 -5,369 -5,336 -5,279 -5,135 -4,999 -4,791
0,55 -5,252 -5,239 -5,220 -5,187 -5,121 -4,986 -4,849 -4,638
0,60 -5,104 -5,091 -5,073 -5,041 -4,976 -4,842 -4,794 -4,492
0,65 -4,956 -4,949 -4,927 -4,896 -4,833 -4,702 -4,565 -4,353
0,70 -4,808 -4,797 -4,781 -4,752 -4,693 -4,566 -4,432 -4,221
0,75 -4,655 -4,646 -4,632 -4,607 -4,554 -4,434 -4,393 -4,095
0,80 -4,494 -4,488 -4,478 -4,459 -4,413 -4,303 -4,178 -3,974
0,85 -4,316 -4,316 -4,312 -4,302 -4,269 -4,173 -4,056 -3,857
0,90 -4,108 -4,118 -4,127 -4,132 -4,119 -4,043 -3,935 -3,744
0,93 -3,953 -3,976 -4,000 -4,020 -4,024 -3,963 -3,863 -3,678
0,95 -3,825 -3,865 -3,904 -3,940 -3,958 -3,910 -3,815 -3,634
0,97 -3,658 -3,732 -3,796 -3,853 -3,890 -3,856 -3,767 -3,591
0,98 -3,544 -3,652 -3,736 -3,806 -3,854 -3,829 -3,743 -3,569
0,99 -3,376 -3,558 -3,670 -3,758 -3,818 -3,801 -3,719 -3,548
1,00 -2,584 -3,441 -3,598 -3,706 -3,782 -3,774 -3,695 -3,526
1,01 -1,796 -3,283 -3,516 -3,652 -3,744 -3,746 -3,671 -3,505
1,02 -1,627 -3,039 -3,422 -3,595 -3,705 -3,718 -3,647 -3,484
1,05 -1,359 -2,034 -3,030 -3,398 -3,583 -3,632 -3,575 -3,420
1,10 -1,120 -1,487 -2,203 -2,965 -3,353 -3,484 -3,453 -3,315
1,15 -0,968 -1,239 -1,719 -2,479 -3,091 -3,329 -3,329 -3,211
1,20 -0,857 -1,076 -1,443 -2,079 -2,801 -3,166 -3,202 -3,107
1,30 -0,698 -0,860 -1,116 -1,560 -2,274 -2,825 -2,942 -2,899
1,40 -0,588 -0,716 -0,915 -1,253 -1,857 -2,486 -2,679 -2,692
1,50 -0,505 -0,611 -0,774 -1,046 -1,549 -2,175 -2,421 -2,486
1,60 -0,440 -0,531 -0,667 -0,894 -1,318 -1,904 -2,177 -2,285
1,70 -0,387 -0,446 -0,583 -0,777 -1,139 -1,672 -1,953 -2,091
1,80 -0,344 -0,413 -0,515 -0,683 -0,996 -1,476 -1,751 -1,908
1,90 -0,307 -0,368 -0,458 -0,606 -0,880 -1,309 -1,571 -1,736
2,00 -0,276 -0,330 -0,411 -0,541 -0,782 -1,167 -1,411 -1,577
2,20 -0,226 -0,269 -0,334 -0,437 -0,629 -0,937 -1,143 -1,295
2,40 -0,187 -0,222 -0,275 -0,359 -0,513 -0,761 -0,929 -1,058
2,60 -0,155 -0,185 -0,228 -0,297 -0,422 -0,621 -0,756 -0,858
2,80 -0,130 -0,154 -0,190 -0,246 -0,348 -0,508 -0,614 -0,689
3,00 -0,109 -0,129 -0,159 -0,205 -0,288 -0,415 -0,495 -0,545
3,50 -0,069 -0,081 -0,099 -0,127 -0,174 -0,239 -0,270 -0,264
4,00 -0,041 -0,048 -0,058 -0,072 -0,095 -0,116 -0,110 -0,061
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
342
ENTALPIA RESIDUA: [h
R
/(RT
C
)]
1
P
R
0,0100 0,0500 0,1000 0,2000 0,4000 0,6000 0,8000 1,0000
T
R
0,30 -11,098 -11,096 -11,095 -11,091 -11,083 -11,076 -11,069 -11,062
0,35 -10,656 -10,655 -10,654 -10, 653 -10,650 -10,646 -10,643 -10,640
0,40 -10,121 -10,121 -10,121 -10,120 -10,121 -10,121 -10,121 -10,121
0,45 -9,515 -9,515 -9,516 9,517 -9,519 -9,521 -9,523 -9,525
0,50 -8,868 -8,869 -8,870 -8,872 -8,876 -8,880 -8,884 -8,888
0,55 -0,080 -8,211 -8,212 -8,215 -8,221 -8,226 -8,232 -8,238
0,60 -0,059 -7,568 -7,570 -7,573 -7,579 -7,585 -7,591 -7,596
0,65 -0,045 -0,247 -6,949 -6,952 -6,959 -6,966 -6,973 -6,980
0,70 -0,034 -0,185 -0,415 -6,360 -6,367 -6,373 -6,381 -6,388
0,75 -0,027 -0,142 -0,306 -5,796 -5,802 -5,809 -5,816 -5,824
0,80 -0,021 -0,110 -0,234 -0,542 -5,266 -5,271 -5,278 -5,285
0,85 -0,017 -0,087 -0,182 -0,401 -4, 753 -4,754 -4,758 -4,763
0,90 -0,014 -0,070 -0,144 -0,308 -0,751 -4,254 -4,248 -4,249
0,93 -0,012 -0,061 -0,126 -0,265 -0,612 -1,236 -3,942 -3,934
0,95 -0,011 -0,056 -0,115 -0,241 -0,542 -0,994 -3,737 -3,712
0,97 -0,010 -0,052 -0,105 -0,219 -0,483 -0,837 -1,616 -3,470
0,98 -0,010 -0,050 -0,101 -0,209 -0,457 -0,776 -1,324 -3,332
0,99 -0,009 -0,048 -0,097 -0,200 -0,433 -0,722 -1,154 -3,164
1,00 -0,009 -0,046 -0,093 -0,191 -0,410 -0,675 -1,034 -2,471
1,01 -0,009 -0,044 -0,089 -0,183 -0,389 -0,632 -0,940 -1,375
1,02 -0,008 -0,042 -0,085 -0,175 -0,370 -0,594 -0,863 -1,180
1,05 -0,007 -0,037 -0,075 -0,153 -0,318 -0,498 -0,691 -0,877
1,10 -0,006 -0,030 -0,061 -0,123 -0,251 -0,381 -0,507 -0,617
1,15 -0,005 -0,025 -0,050 -0,099 -0,199 -0,296 -0,385 -0,459
1,20 -0,004 -0,020 -0,040 -0,080 -0,158 -0,232 -0,297 -0,349
1,30 -0,003 -0,013 -0,026 -0,052 -0,100 -0,142 -0,177 -0,203
1,40 -0,002 -0,008 -0,016 -0,032 -0,060 -0,083 -0,100 -0,111
1,50 -0,001 -0,005 -0,009 -0,018 -0,032 -0,042 -0,048 -0,049
1,60 -0,000 -0,002 -0,004 -0,007 -0,012 -0,013 -0,011 -0,005
1,70 -0,000 -0,000 -0,000 -0,000 0,003 0,009 0,017 0,027
1,80 0,000 0,001 0,003 0,006 0,015 0,025 0,037 0,051
1,90 0,001 0,003 0,005 0,011 0,023 0,037 0,053 0,070
2,00 0,001 0,003 0,007 0,015 0,030 0,047 0,065 0,085
2,20 0,001 0,005 0,010 0,020 0,040 0,062 0,083 0, 106
2,40 0,001 0,006 0,012 0,023 0,047 0,071 0,095 0,120
2,60 0,001 0,006 0,013 0,026 0,052 0,078 0,104 0,130
2,80 0,001 0,007 0,014 0,028 0,055 0,082 0,110 0,137
3,00 0,001 0,007 0,014 0,029 0,058 0,086 0,114 0,142
3,50 0,002 0,008 0,016 0,031 0,062 0,092 0,122 0,152
4,00 0,002 0,008 0,016 0,032 0,064 0,096 0,127 0,158
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
343
P
R
1,0000 1,2000 1,5000 2,0000 3,0000 5,0000 7,0000 10,000
T
R
0,30 -11,062 -11,055 -11,044 -11,027 -10,992 -10,935 -10,872 -10,781
0,35 -10,640 -10,637 -10,632 -10,624 -10,609 -10,581 -10,554 -10,529
0,40 -10,121 -10,121 -10,121 -10,122 -10,123 -10,128 -10,135 -10,150
0,45 -9,525 -9,527 -9,531 -9,537 -9,549 -9,576 -9,611 -9,663
0,50 -8,888 -8,892 -8,899 -8,909 -8,932 -8,978 -9,030 -9,111
0,55 -8,238 -8,243 -8,252 -8,267 -8,298 -8,360 -8,425 -8,531
0,60 -7,596 -7,603 -7,614 -7,632 -7,669 -7,745 -7,824 -7,950
0,65 -6,980 -6,987 -6,997 -7,017 -7,059 -7,147 -7,239 -7,381
0,70 -6,388 -6,395 -6,407 -6,429 -6,475 -6,574 -6,677 -6,837
0,75 -5,824 -5,832 -5,845 -5,868 -5,918 -6,027 -6,142 -6,318
0,80 -5,285 -5,293 -5,306 -5,330 -5,385 -5,506 -5,632 -5,824
0,85 -4,763 -4,771 -4,784 -4,810 -4,872 -5,000 -5,149 -5,358
0,90 -4,249 -4,255 -4,268 -4,298 -4,371 -4,530 -4,688 -4,916
0,93 -3,934 -3,937 -3,951 -3,987 -4,073 -4,251 -4,422 -4,662
0,95 -3,712 -3,713 -3,730 -3,773 -3,873 -4,068 -4,248 -4,497
0,97 -3,470 -3,467 -3,492 -3,551 -3,670 -3,885 -4,077 -4,336
0,98 -3,332 -3,327 -3,363 -3,434 -3,568 -3,795 -3,992 -4,257
0,99 -3,164 -3,164 -3,223 -3,313 -3,464 -3,705 -3,909 -4,178
1,00 -2,471 -2,952 -3,065 -3,186 -3,358 -1615 -3,825 -4,100
1,01 -1,375 -2,595 -2,880 -3,051 -3,251 -3,525 -3,742 -4,023
1,02 -1,180 -1,723 -2,650 -2,906 -3,142 -3,435 -3,661 -3,947
1,05 -0,877 -0,878 -1,496 -2,381 -2,800 -3,167 -3,418 -3,722
1,10 -0,617 -0,673 -0,617 -1,261 -2,167 -2,720 -3,023 -3,362
1,15 -0,459 -0,503 -0,487 -0,604 -1,497 -2,275 -2,641 -3,019
1,20 -0,349 -0,381 -0,381 -0,361 -0,934 -1,840 -2,273 -2,692
1,30 -0,203 -0,218 -0,218 -0,178 -0,300 -1,066 1,592 -2,086
1,40 -0,111 -0,115 -0,128 -0,070 -0,044 -0,504 -1,012 -1,547
1,50 -0,049 -0,046 -0,032 0,008 0,078 -0,142 -0,556 -1,080
1,60 -0,005 0,004 0,023 0,065 0,151 0,082 -0,217 -0,689
1,70 0,027 0,040 0,063 0,109 0,202 0,223 0,028 -0,369
1,80 0,051 0,067 0,094 0,143 0,241 0,317 0,203 -0,112
1,90 0,070 0,088 0,117 0,169 0,271 0,381 0,330 0,092
2,00 0,085 0,105 0,136 0,190 0,295 0,428 0,424 0,255
2,20 0,106 0,128 0,163 0,221 0,331 0,493 0,551 0,489
2,40 0,120 0,144 0,181 0,242 0,356 0,535 0,631 0,645
2,60 0,130 0,156 0,194 0,257 0,376 0,567 0,687 0,754
2,80 0,137 0,164 0,204 0,269 0,391 0,591 0,729 0,836
3,00 0,142 0,170 0,211 0,278 0,403 0,611 0,763 0,899
3,50 0,152 0,181 0,224 0,294 0,425 0,650 0,827 1,015
4,00 0,158 0,188 0,233 0,306 0,442 0,680 0,874 1,097
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
344
ENTROPIA RESIDUA: [s
R
/R]
0
P
R
0,0100 0,0500 0,1000 0,2000 0,4000 0,6000 0,8000 1,0000
T
R
0,30 -11,614 -10,008 -9,319 -8,635 -7,961 -7,574 -7,304 -7,099
0,35 -11,185 -9,579 -8, 890 -8,205 -7,529 -7,140 -6,869 -6,663
0,40 -10,802 -9,196 -8,506 -7,821 -7,144 -6,755 -6,483 -6,275
0,45 -10,453 -8,847 -8,157 -7,472 -6,794 -6,404 -6,132 -5,924
0,50 -10,137 -8,531 -7,841 -7,156 -6,479 -6,089 -5,816 -5,608
0,55 -0,038 -8,245 -7,555 -6,870 -6,193 -5,803 -5,531 -5,324
0,60 -0,029 -7,983 -7,294 -6,610 -5,933 -5,544 -5,273 -5,066
0,65 -0,023 -0,122 -7,052 -6,368 -5,694 -5,306 -5,036 -4,830
0,70 -0,018 -0,096 -0,206 -6,140 -5,467 -5,082 -4,814 -4,610
0,75 -0,015 -0,078 -0,164 -5,917 -5,248 -4,866 -4,600 -4,399
0,80 -0,013 -0,064 -0,134 -0,294 -5,026 -4,694 -4,388 -4,191
0,85 -0,011 -0,054 -0,111 -0,239 -4,785 -4,418 -4,166 -3,976
0,90 -0,009 -0,046 -0,094 -0,199 -0,463 -4,145 -3,912 -3,738
0,93 -0,008 -0,042 -0,085 -0,179 -0,408 -0,750 -3,723 -3,569
0,95 -0,008 -0,039 -0,080 -0,168 -0,377 -0,671 -3,556 -3,433
0,97 -0,007 -0,037 -0,075 -0,157 -0,350 -0,607 -1,056 -3,259
0,98 -0,007 -0,036 -0,073 -0,153 -0,337 -0,580 -0,971 -3,142
0,99 -0,007 -0,035 -0,071 -0,148 -0,326 -0,555 -0,903 -2,972
1,00 -0,007 -0,034 -0,069 -0,144 -0,315 -0,532 -0,847 -2,178
1,01 -0,007 -0,033 -0,067 -0,139 -0,304 -0,510 -0,799 -1,391
1,02 -0,006 -0,032 -0,065 -0,135 -0,294 -0,491 -0,757 -1,225
1,05 -0,006 -0,030 -0,060 -0,124 -0,267 -0,439 -0,656 -0,965
1,10 -0,005 -0,026 -0,053 -0,108 -0,230 -0,371 0,537 -0,742
1,15 -0,005 -0,023 -0,047 -0,096 -0,201 -0,319 -0,452 -0,607
1,20 -0,004 -0,021 -0,042 -0,085 -0,177 -0,277 -0,389 -0,512
1,30 -0,003 -0,017 -0,033 -0,068 -0,140 -0,217 -0,298 -0,385
1,40 -0,003 -0,014 -0,027 -0,056 -0,114 -0,174 -0,237 -0,303
1,50 -0,002 -0,01,1 -0,023 -0,046 -0,094 -0,143 -0,194 -0,246
1,60 -0,002 -0,010 -0,019 -0,039 -0,079 -0,120 -0,162 -0,204
1,70 -0,002 -0,008 -0,017 -0,033 -0,067 -0,102 -0,137 -0,172
1,80 -0,001 -0,007 -0,014 -0,029 -0,058 -0,088 -0,117 -0,147
1,90 -0,001 -0,006 -0,013 -0,025 -0,051 -0,076 -0,102 -0,127
2,00 -0,001 -0,006 -0,011 -0,022 -0,044 -0,067 -0,089 -0,111
2,20 -0,001 -0,004 -0,009 -0,018 -0,035 -0,053 -0,070 -0,087
2,40 -0,001 -0,004 -0,007 -0,014 -0,028 -0,042 -0,056 -0,070
2,60 -0,001 -0,003 -0,006 -0,012 -0,023 -0,035 -0,046 -0,058
2,80 -0,000 -0,002 -0,005 -0,010 -0,020 -0,029 -0,039 -0,048
3,00 -0,000 -0,002 -0,004 -0,008 -0,017 -0,025 -0,033 -0,041
3,50 -0,000 -0,001 -0,003 -0,006 -0,012 -0,017 -0,023 -0,029
4,00 -0,000 -0,001 -0,002 -0,004 -0,009 -0,013 -0,017 -0,021
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
345
P
R
1,0000 1,2000 1,5000 2,0000 3,0000 5,0000 7,0000 10,000
T
R
0,30 -7,099 -6,935 6,740 -6,497 -6,180 -5,847 -5,683 -5,578
0,35 -6,663 -6,497 -6,2,99 -6,052 -5,728 -5,376 -5,194 -5,060
0,40 -6,275 -6,109 -5,909 -5,66,0 -5,330 -4,967 -4,772 -4,619
0,45 -5,924 -5,757 -5,557 -5,306 -4,974 -4,603 -4,401 -4,234
0,50 -5,608 -5,441 -5,240 -4,989 -4,656 -4,282 -4,074 -3,899
0,55 -5,324 -5,157 -4,956 -4,706 -4,373 -3,998 -3,788 -3,607
0,60 -5,066 -4,900 -4,700 -4,451 -4,120 -3,747 -3,537 -3,353
0,65 -4,830 -4,665 -4,467 -4,220 -3,892 -3,523 -3,315 -3,131
0,70 -4,610 -4,446 -4,250 -4,007 -3,684 -3,322 -3,117 -2,935
0,75 -4,399 -4,238 -4,045 -3,807 -3,491 -3,138 -2,939 -2,761
0,80 -4,191 -4,034 -3,846 -3,615 -3,310 -2,970 -2,777 -2,605
0,85 -3,976 -3,825 -3,646 -3,425 -3,135 -2,812 -2,629 -2,463
0,90 -3,738 -3,599 -3,434 -3,231 -2,964 -2,663 -2,491 -2,334
0,93 -3,569 -3,444 -3,295 -3,108 -2,860 -2,577 -2,412 -2,262
0,95 -3,433 -3,326 -3,193 -3,023 -2,790 -2,520 -2,362 -2,215
0,97 -3,259 -3,188 -3,081 -2,932 -2,719 -2,463 -2,312 -2,170
0,98 -3,142 -3,106 -3,019 -2,884 -2,682 -2,436 -2,287 -2,148
0,99 -2,972 -3,010 -2,953 -2,835 -2,646 -2,408 -2,263 -2,126
1,00 -2,178 -2,893 -2,879 -2,784 -2,609 -2,380 -2,239 -2,105
1,01 -1,391 -2,736 -2,798 -2,730 -2,571 -2,352 -2,215 -2,083
1,02 -1,225 -2,495 -2,706 -2,673 -2,533 -2,325 -2,191 -2,062
1,05 -0,965 -1,523 -2328 -2,483 -2,415 -2,242 -2,121 -2,001
1,10 -0,742 -1,012 -1,557 -2,081 -2,202 -2,104 -2,007 -1,903
1,15 -0,607 -0,790 -1,126 -1,649 -1,968 -1,966 -1,897 -1,810
1,20 -0,512 -0,651 -0,890 -1,308 -1,727 -1,827 -1,789 -1,722
1,30 -0,385 -0,478 -0,628 -0,891 -1,299 -1,554 -1,581 -1,556
1,40 -0,303 -0,375 -0,478 -0,663 -0,990 -1,303 -1,386 -1,402
1,50 -0,246 -0,299 -0,381 -0,520 -0,777 -1,088 -1,208 -1,260
1,60 -0,204 -0,247 -0,312 -0,421 -0,628 -0,913 -1,050 -1,130
1,70 -0,172 -0,208 -0,261 -0,350 -0,519 -0,773 -0,915 -1,013
1,80 -0,147 -0,177 -0,222 -0,296 -0,438 -0,661 -0,799 -0,908
1,90 -0,127 -0,153 -10,191 -0,255 -0,375 -0,570 -0,702 -0,815
2,00 -0,111 -0,134 -0,167 -0,221 -0,625 -0,497 -0,620 -0,733
2,20 -0,087 -0,105 -0,130 -0,172 -0,251 -0,388 -0,492 -0,599
2,40 -0,070 -0,084 -0,104 -0,138 -0,201 -0,311 -0,399 -0,496
2,60 -0,058 -0,069 -0,086 -0,113 -0,164 -0,255 -0,329 -0,416
2,80 -0,048 -0,058 -0,072 -0,094 -0,137 -0,213 -0,277 -0,353
3,00 -0,041 -0,049 -0,061 -0,080 -0,116 -0,181 -0,236 -0,303
3,50 -0,029 -0,034 -0,042 -0,056 -0,081 -0,126 -0,166 -0,216
4,00 -0,021 -0,025 -0,031 -0,041 -0,059 -0,093 -0,123 -0,162
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
346
ENTROPIA RESIDUA: [s
R
/R]
1
P
R
0,0100 0,0500 0,1000 0,2000 0,4000 0,6000 0,8000 1,0000
T
R
0,30 -16,782 -16,774 -16,764 -16,744 -16,705 -16,665 -16,626 -16,586
0,35 -15,413 -15,408 -15,401 -15,387 -15,359 -15,333 -15,305 -15,278
0,40 -13,990 -13,986 -13,981 -13,972 -13,953 -13,934 -13,915 -13,896
0,45 -12,564 -12,561 -12,558 -12,551 -12,537 -12,523 -12,509 -12,496
0,50 -11,202 -11,200 -11,197 -11,092 -11,082 -11,172 -11,162 -11,153
0,55 -0,115 -9,948 -9,946 -9,942 -9,935 -9,928 -9,921 -9,914
0,60 -0,078 -8,828 -8,826 -8,823 -8,817 -8,811 -8,806 -8,799
0,65 -0,055 -0,309 -7,832 -7,829 -7,824 -7,819 -7,815 -7,510
0,70 -0,040 -0,216 -0,491 -6,951 6,945 -6,941 -6,937 -6,933
0,75 -0,029 -0,156 -0,340 -6,173 -6,167 -6,162 -6,158 -6,155
0,80 -0,022 -0,116 -0,246 -0,578 -5,475 -5,468 -5,462 -5,458
0,85 -0,017 -0,088 -0,183 -0,400 -4,853 -4,841 -4,832 -4,826
0,90 -0,013 -0,068 -0,140 -0,301 -0,744 -4,269 -4,249 -4,238
0,93 -0,011 -0,058 -0,120 -0,254 -0,593 -1,219 -3,914 -3,894
0,95 -0,010 -0,053 -0,109 -0,228 -0,517 -0,961 -3,697 -3,658
0,97 -0,010 -0,048 -0,099 -0,206 -0,456 -0,797 -1,570 -3,406
0,98 -0,009 -0,046 -0,094 -0,196 -0,429 -0,734 -1,270 -3,264
0,99 -0,009 -0,044 -0,090 -0,186 -0,405 -0,680 -1,098 -3,093
1,00 -0,008 -0,042 -0,086 -0,177 -0,382 -0,632 -0,977 -2,399
1,01 -0,008 -0,040 -0,082 -0,169 -0,361 -0,590 -0,883 -1,306
1,02 -0,008 -0,039 -0,078 -0,161 -0,342 -0,552 -0,807 -1,113
1,05 -0,007 -0,034 -0,069 -0,140 -0,292 -0,460 -0,642 -0,820
1,10 -0,005 -0,028 -0,055 -0,112 -0,229 -0,350 -0,470 -0,577
1,15 -0,005 -0,023 -0,045 -0,091 -0,183 -0,275 -0,361 -0,437
1,20 -0,004 -0,019 -0,037 -0,075 -0,149 -0,220 -0,286 -0,343
1,30 -0,003 -0,013 -0,026 -0,052 -0,102 -0,148 -0,190 -0,226
1,40 -0,002 -0,010 -0,019 -0,037 -0,072 -0,104 -0,133 -0,158
1,50 -0,001 -0,007 -0,014 -0,027 -0,053 -0,076 -0,097 -0,115
1,60 -0,001 -0,005 -0,011 -0,021 -0,040 -0,057 -0,073 -0,086
1,70 -0,001 -0,004 -0,008 -0,016 -0,031 -0,044 -0,056 -0,067
1,80 -0,001 -0,003 -0,006 -0,013 -0,024 -0,035 -0,044 -0,053
1,90 -0,001 -0,003 -0,005 -0,010 -0,019 -0,028 -0,036 -0,043
2,00 -0,000 -0,002 -0,004 -0,008 -0,016 -0,023 -0,029 -0,035
2,20 -0,000 -0,001 -0,003 -0,006 -0,011 -0,016 0,021 0,025
2,40 -0,000 -0,001 -0,002 -0,004 -0,008 -0,012 -0,015 -0,019
2,60 -0,000 -0,001 -0,002 -0,003 -0,006 -0,009 -0,012 -0,015
2,80 -0,000 -0,001 -0,001 -0,003 -0,005 -0,008 -0,010 -0,012
3,00 -0,000 -0,001 -0,001 -0,002 -0,004 -0,006 -0,008 -0,010
3,50 -0,000 -0,000 -0,001 -0,001 -0,003 -0,004 -0,006 -0,007
4,00 -0,000 -0,000 -0,001 -0,001 -0,002 -0,003 -0,005 -0,006
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
347
P
R
1,0000 1,2000 1,5000 2,0000 3,0000 5,0000 7,0000 10,000
T
R
0,30 -16,586 -16,547 -16,488 -16,390 -16,195 -15,837 -15,468 -14,925
0,35 -15,278 -15,251 -15,211 -15,144 -15,011 -14,751 -14,496 -14,153
0,40 -13,896 -13,877 -13,849 -13,803 -13,714 -13,541 -13,376 -13,144
0,45 -12,496 -12,482 -12,462 -12,430 -12,367 -12,248 -12,145 -11,999
0,50 -11,153 -11,143 -11,129 -11,107 -11,063 -10,985 -10,920 -10,836
0,55 -9,914 -9,907 -9,897 -9,882 -9,853 -9,806 -9,769 -9,732
0,60 -8,799 -8,794 -8,787 -8,777 -8,760 -8,736 -8,723 -8,720
0,65 -7,810 -7,807 -7,801 -7,794 -7,784 -7,779 -7,785 -7,811
0,70 -6,933 -6,930 -6,926 -6,922 -6,919 -6,929 -6,952 -7,002
0,75 -6,155 -6,152 -6,149 -6,147 -6,149 -6,174 -6,213 -6,285
0,80 -5,458 -5,455 -5,453 -5,452 -5,461 -5,501 -5,555 -5,648
0,85 -4,826 -4,822 -4,820 -4,822 -4,839 -4,898 -4,969 -5,082
0,90 -4,238 -4,232 -4,230 -4,236 -4,267 -4,351 -4,442 -4,578
0,93 -3,894 -3,885 -3,884 -3,896 -3,941 -4,046 -4,151 -4,300
0,95 -3,658 -3,647 -3,648 -3,669 -3,728 -3,851 -3,966 -4,125
0,97 -3,406 -3,391 -3,401 -3,437 -3,517 -3,661 -3,788 -3,957
0,98 -3,264 -3,247 -3,268 -3318 -3,412 -3,569 -3,701 -3,875
0,99 -3,093, -3,082 -3,126 -3,195 -3,306 -3,477 -3,616 -3,796
1,00 -2,399 -2,868 -2,967 -9,067 -3,200 -3,387 -3,532 -3,717
1,01 -1,306 -2,513 -2,784 -2,933 -3,094 -3,297 -3,450 -3,640
1,02 -1,113 -1,655 -2157 -2,790 -2,986 -3,209 -3,369 -3,565
1,05 -0,820 -0,831 -1,443 -2,283 -2,655 -2,949 -3,134 -3,348
1,10 -0,577 -0,640 -0,618 -1,241 -2,067 -2,534 -2,767 -3,013
1,15 -0,437 -0,489 -0,502 -0,654 -1,471 -2,138 -2,428 -2,708
1,20 -0,343 -0,385 -0,412 -0,447 -0,991 -1,767 -2,115 -2,430
1,30 -0,226 -0,254 -0,282 -0,300 -0,481 -1,147 -1,569 -1,944
1,40 -0,158 -0,178 -0,200 -0,220 -0,290 -0,730 -1,138 -1,544
1,50 -0,115 -0,130 -0,147 -0,166 -0,206 -0,479 -0,823 -1,222
1,60 -0,086 -0,098 -0,112 -0,129 -0,159 -0,334 -0,604 -0,969
1,70 -0,067 -0,076 -0,087 -0,102 -0,127 -0,248 -0,456 -0,775
1,80 -0,053 -0,060 -0,070 -0,083 -0,105 -0,195 -0,355 -0,628
1,90 -0,043 -0,049 -0,057 -0,069 -0,089 -0,160 -0,286 -0,518
2,00 -0,035 -0,040 -0,048 -0,058 -0,077 -0,136 -0,238 -0,434
2,20 -0,025 -0,029 -0,035 -0,043 -0,060 -0,105 -0,178 -0,322
2,40 -0,019 -0,022 -0,027 -0,034 -0,048 -0,086 -0,143 -0,254
2,60 -0,015 -0,018 -0,021 -0,028 -0,041 -0,074 -0,120 -0,210
2,80 -0,012 -0,014 -0,018 -0,023 -0,025 -0,065 -0,104 -0,180
3,00 -0,010 -0,012 -0,015 -0,020 -0,031 -0,058 -0,093 -0,158
3,50 -0,007 -0,009 -0,011 -0,015 -0,024 -0,046 -0,073 -0,122
4,00 -0,006 -0,007 -0,009 -0,012 -0,020 -0,038 -0,060 -0,100
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
348
COEFFICIENTE DI FUGACIT:
0
P
R
0,0100 0,0500 0,1000 0,2000 0,4000 0,6000 0,8000 1,0000
T
R
0,30 0,0002 0,0000 0,0000 0,0000 0,0000 0,0000 0,0000 0,0000
0,35 0,0034 0,0007 0,0003 0,0002 0,0001 0,0001 0,0001 0,0000
0,40 0,0272 0,0055 0,0028 0,0014 0,0007 0,0005 0,0004 0,0003
0,45 0,1321 0,0266 0,0135 0,0069 0,0036 0,0025 0,0020 0,0016
0,50 0,4529 0,0912 0,0461 0,0235 0,0122 0,0085 0,0067 0,0055
0,55 0,9817 0,2432 0,1227 0,0625 0,0325 0,0225 0,0176 0,0146
0,60 0,9840 0,5383 0,2716 0,1384 0,0718 0,0497 0,0386 0,0321
0,65 0,9886 0,9419 0,5212 0,2655 0,1374 0,0948 0,0738 0,0611
0,70 0,9908 0,9528 0,9057 0,4560 0,2360 0,1626 0,1262 0,1045
0,75 0,9931 0,9616 0,9226 0,7178 0,3715 0,2559 0,1982 0,1641
0,80 0,9931 0,9683 0,9354 0,8730 0,5445 0,3750 0,2904 0,2404
0,85 0,9954 0,9727 0,9462 0,8933 0,7534 0,5188 0,4018 0,3319
0,90 0,9954 0,9772 0,9550 0,9099 0,8204 0,6823 0,5297 0,4375
0,93 0,9954 0,9795 0,9594 0,9183 0,8375 0,7551 0,6109 0,5058
0,95 0,9954 0,9817 0,9616 0,9226 0,8472 0,7709 0,6668 0,5521
0,97 0,9954 0,9817 0,9638 0,9268 0,8570 0,7852 0,7112 0,5984
0,98 0,9954 0,9817 0,9638 0,9290 0,8610 0,7925 0,7211 0,6223
0,99 0,9977 0,9840 0,9661 0,9311 0,8650 0,7980 0,7295 0,6442
1,00 0,9977 0,9840 0,9661 0,9333 0,8690 0,8035 0,7379 0,6668
1,01 0,9977 0,9840 0,9683 0,9354 0,8730 0,8110 0,7464 0,6792
1,02 0,9977 0,9840 0,9683 0,9376 0,8770 0,8166 0,7551 0,6902
1,05 0,9977 0,9863 0,9705 0,9441 0,8872 0,8318 0,7762 0,7194
1,10 0,9977 0,9886 0,9750 0,9506 0,9016 0,8531 0,8072 0,7586
1,15 0,9977 0,9886 0,9795 0,9572 0,9141 0,8730 0,8318 0,7907
1,20 0,9977 0,9908 0,9817 0,9616 0,9247 0,8892 0,8531 0,8166
1,30 0,9977 0,9931 0,9863 0,9705 0,9419 0,9141 0,8872 0,8590
1,40 0,9977 0,9931 0,9886 0,9772 0,9550 0,9333 0,9120 0,8892
1,50 1,0000 0,9954 0,9908 0,9817 0,9638 0,9462 0,9290 0,9141
1,60 1,0000 0,9954 0,9931 0,9863 0,9727 0,9572 0,9441 0,9311
1,70 1,0000 0,9977 0,9954 0,9886 0,9772 0,9661 0,9550 0,9462
1,80 1,0000 0,9977 0,9954 0,9908 0,9817 0,9727 0,9661 0,9572
1,90 1,0000 0,9977 0,9954 0,9931 0,9863 0,9795 0,9727 0,9661
2,00 1,0000 0,9977 0,9977 0,9954 0,9886 0,9840 0,9795 0,9727
2,20 1,0000 1,0000 0,9977 0,9977 0,9931 0,9908 0,9886 0,9840
2,40 1,0000 1,0000 1,0000 0,9977 0,9977 0,9954 0,9931 0,9931
2,60 1,0000 1,0000 1,0000 1,0000 1,0000 0,9977 0,9977 0,9977
2,80 1,0000 1,0000 1,0000 1,0000 1,0000 1,0000 1,0023 1,0023
3,00 1,0000 1,0000 1,0000 1,0000 1,0023 1,0023 1,0046 1,0046
3,50 1,0000 1,0000 1,0000 1,0023 1,0023 1,0046 1,0069 1,0093
4,00 1,0000 1,0000 1,0000 1,0023 1,0046 1,0069 1,0093 1,0116
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
349
P
R
1,0000 1,2000 1,5000 2,0000 3,0000 5,0000 7,0000 10,000
T
R
0,30 0,0000 0,0000 0,0000 0,0000 0,0000 0,0000 0,0000 0,0000
0,35 0,0000 0,0000 0,0000 0,0000 0,0000 0,0000 0,0000 0,0000
0,40 0,0003 0,0003 0,0003 0,0002 0,0002 0,0002 0,0002 0,0003
0,45 0,0016 0,0014 0,0012 0,0010 0,0008 0,0008 0,0009 0,0012
0,50 0,0055 0,0048 0,0041 0,0034 0,0028 0,0025 0,0027 0,0034
0,55 0,0146 0,0127 0,0107 0,0089 0,0072 0,0063 0,0066 0,0080
0,60 0,0321 0,0277 0,0234 0,0193 0,0154 0,0132 0,0135 0,0160
0,65 0,0611 0,0527 0,0445 0,0364 0,0289 0,0244 0,0245 0,0282
0,70 0,1045 0,0902 0,0759 0,0619 0,0488 0,0406 0,0402 0,0453
0,75 0,1641 0,1413 0,1188 0,0966 0,0757 0,0625 0,0610 0,0673
0,80 0,2404 0,2065 0,1738 0,1409 0,1102 0,0899 0,0867 0,0942
0,85 0,3319 0,2858 0,2399 0,1945 0,1517 0,1227 0,1175 0,1256
0,90 0,4375 0,3767 0,3162 0,2564 0,1995 0,1607 0,1524 0,1611
0,93 0,5058 0,4355 0,3656 0,2972 0,2307 0,1854 0,1754 0,1841
0,95 0,5521 0,4764 0,3999 0,3251 0,2523 0,2028 0,1910 0,2000
0,97 0,5984 0,5164 0,4345 0,3532 0,2748 0,2203 0,2075 0,2163
0,98 0,6223 0,5370 0,4529 0,3681 0,2864 0,2296 0,2158 0,2244
0,99 0,6442 0,5572 0,4699 0,3828 0,2978 0,2388 0,2244 0,2328
1,00 0,6668 0,5781 0,4875 0,3972 0,3097 0,2483 0,2328 0,2415
1,01 0,6792 0,5970 0,5047 0,4121 0,3214 0,2576 0,2415 0,2500
1,02 0,6902 0,6166 0,5224 0,4266 0,3334 0,2673 0,2506 0,2582
1,05 0,7194 0,6607 0,5728 0,4710 0,3690 0,2958 0,2773 0,2844
1,10 0,7586 0,7112 0,6412 0,5408 0,4285 0,3451 0,3228 0,3296
1,15 0,7907 0,7499 0,6918 0,6026 0,4875 0,3954 0,3690 0,3750
1,20 0,8166 0,7834 0,7328 0,6546 0,5420 0,4446 0,4150 0,4198
1,30 08590 0,8318 0,7943 0,7345 0,6383 0,5383 0,5058 0,5093
1,40 0,8892 0,8690 0,8395 0,7925 0,7145 0,6237 0,5902 0,5943
1,50 0,9141 0,8974 0,8730 0,8375 0,7745 0,6966 0,6668 0,6714
1,60 0,9311 0,9183 0,8995 0,8710 0,8222 0,7586 0,7328 0,7430
1,70 0,9462 0,9354 0,9204 0,8995 0,8610 0,8091 0,7907 0,8054
1,80 0,9572 0,9484 0,9376 0,9204 0,8913 0,8531 0,8414 0,8590
1,90 0,9661 0,9594 0,9506 0,9376 0,9162 0,8872 0,8831 0,9057
2,00 0,9727 0,9683 0,9616 0,9528 0,9354 0,9183 0,9183 0,9462
2,20 0,9840 0,9817 0,9795 0,9727 0,9661 0,9616 0,9727 1,0093
2,40 0,9931 0,9908 0,9908 0,9886 0,9863 0,9931 1,0116 1,0568
2,60 0,9977 0,9977 0,9977 0,9977 1,0023 1,0162 1,0399 1,0889
2,80 1,0023 1,0023 1,0046 1,0069 1,0116 1,0328 1,0593 1,1117
3,00 1,0046 1,0069 1,0069 1,0116 1,0209 1,0423 1,0740 1,1298
3,50 1,0093 1,0116 1,0139 1,0186 1,0304 1,0593 1,0914 1,1508
4,00 1,0116 1,0139 1,0162 1,0233 1,0375 1,0666 1,0990 1,1588
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
350
COEFFICIENTE DI FUGACIT:
1
P
R
0,0100 0,0500 0,1000 0,2000 0,4000 0,6000 0,8000 1,0000
T
R
0,30 0,0000 0,0000 0,0000 0,0000 0,0000 0,0000 0,0000 0,0000
0,35 0,0000 0,0000 0,0000 0,0000 0,0000 0,0000 0,0000 0,0000
0,40 0,0000 0,0000 0,0000 0,0000 0,0000 0,0000 0,0000 0,0000
0,45 0,0002 0,0002 0,0002 0,0002 0,0002 0,0002 0,0002 0,0002
0,50 0,0014 0,0014 0,0014 0,0014 0,0014 0,0014 0,0013 0,0013
0,55 0,9705 0,0069 0,0068 0,0068 0,0066 0,0065 0,0064 0,0063
0,60 0,9795 0,0227 0,0226 0,0223 0,0220 0,0216 0,0213 0,0210
0,65 0,9863 0,9311 0,0572 0,0568 0,0559 0,0551 0,0543 0,0535
0,70 0,9908 0,9528 0,9036 0,1182 0,1163 0,1147 0,1131 0,1116
0,75 0,9931 0,9683 0,9332 0,2112 0,2078 0,2050 0,2022 0,1994
0,80 0,9954 0,9772 0,9550 0,9057 0,3302 0,3257 0,3212 0,3168
0,85 0,9977 0,9863 0,9705 0,9375 0,4774 0,4708 0,4654 0,4590
0,90 0,9977 0,9908 0,9795 0,9594 0,9141 0,6323 0,6250 0,6165
0,93 0,9977 0,9931 0,9840 0,9705 0,9354 0,8953 0,7227 0,7144
0,95 0,9977 0,9931 0,9885 0,9750 0,9484 0,9183 0,7888 0,7797
0,97 1,0000 0,9954 0,9908 0,9795 0,9594 0,9354 0,9078 0,8413
0,98 1,0000 0,9954 0,9908 0,9817 0,9638 0,9440 0,9225 0,8729
0,99 1,0000 0,9954 0,9931 0,9840 0,9683 0,9528 0,9332 0,9036
1,00 1,0000 0,9977 0,9931 0,9863 0,9727 0,9594 0,9440 0,9311
1,01 1,0000 0,9977 0,9931 0,9885 0,9772 0,9638 0,9528 0,9462
1,02 1,0000 0,9977 0,9954 0,9908 0,9795 0,9705 0,9616 0,9572
1,05 1,0000 0,9977 0,9977 0,9954 0,9885 0,9863 0,9840 0,9840
1,10 1,0000 1,0000 1,0000 1,0000 1,0023 1,0046 1,0093 1,0163
1,15 1,0000 1,0000 1,0023 1,0046 1,0116 1,0186 1,0257 1,0375
1,20 1,0000 1,0023 1,0046 1,0069 1,0163 1,0280 1,0399 1,0544
1,30 1,0000 1,0023 1,0069 1,0116 1,0257 1,0399 1,0544 1,0716
1,40 1,0000 1,0046 1,0069 1,0139 1,0304 1,0471 1,0642 1,0815
1,50 1,0000 1,0046 1,0069 1,0163 1,0328 1,0496 1,0666 1,0865
1,60 1,0000 1,0046 1,0069 1,0163 1,0328 1,0496 1,0691 1,0865
1,70 1,0000 1,0046 1,0093 1,0163 1,0328 1,0496 1,0691 1,0865
1,80 1,0000 1,0046 1,0069 1,0163 1,0328 1,0496 1,0666 1,0840
1,90 1,0000 1,0046 1,0069 1,0163 1,0328 1,0496 1,0666 1,0815
2,00 1,0000 1,0046 1,0069 1,0163 1,0304 1,0471 1,0642 1,0815
2,20 1,0000 1,0046 1,0069 1,0139 1,0304 1,0447 1,0593 1,0765
2,40 1,0000 1,0046 1,0069 1,0139 1,0280 1,0423 1,0568 1,0716
2,60 1,0000 1,0023 1,0069 1,0139 1,0257 1,0399 1,0544 1,0666
2,80 1,0000 1,0023 1,0069 1,0116 1,0257 1,0375 1,0496 1,0642
3,00 1,0000 1,0023 1,0069 1,0116 1,0233 1,0352 1,0471 1,0593
3,50 1,0000 1,0023 1,0046 1,0023 1,0209 1,0304 1,0423 1,0520
4,00 1,0000 1,0023 1,0046 1,0093 1,0186 1,0280 1,0375 1,0471
Renato Rota - Fondamenti di Termodinamica dellIngegneria Chimica
351
P
R
1,0000 1,2000 1,5000 2,0000 3,0000 5,0000 7,0000 10,000
T
R
0,30 0,0000 0,0000 0,0000 0,0000 0,0000 0,0000 0,0000 0,0000
0,35 0,0000 0,0000 0,0000 0,0000 0,0000 0,0000 0,0000 0,0000
0,40 0,0000 0,0000 0,0000 0,0000 0,0000 0,0000 0,0000 0,0000
0,45 0,0002 0,0002 0,0002 0,0002 0,0001 0,0001 0,0001 0,0001
0,50 0,0013 0,0013 0,0013 0,0012 0,0011 0,0009 0,0008 0,0006
0,55 0,0063 0,0062 0,0061 0,0058 0,0053 0,0045 0,0039 0,0031
0,60 0,0210 0,0207 0,0202 0,0194 0,0179 0,0154 0,0133 0,0108
0,65 0,0536 0,0527 0,0516 0,0497 0,0461 0,0401 0,0350 0,0289
0,70 0,1117 0,1102 0,1079 0,1040 0,0970 0,0851 0,0752 0,0629
0,75 0,1995 0,1972 0,1932 0,1871 0,1754 0,1552 0,1387 0,1178
0,80 0,3170 0,3133 0,3076 0,2978 0,2812 0,2512 0,2265 0,1954
0,85 0,4592 0,4539 0,4457 0,4325 0,4093 0,3698 0,3365 0,2951
0,90 0,6166 0,6095 0,5998 0,5834 0,5546 0,5058 0,4645 0,4130
0,93 0,7145 0,7063 0,6950 0,6761 0,6457 0,5916 0,5470 0,4898
0,95 0,7798 0,7691 0,7568 0,7379 0,7063 0,6501 0,6026 0,5432
0,97 0,8414 0,8318 0,8185 0,7998 0,7656 0,7096 0,6607 0,5984
0,98 0,8730 0,8630 0,8492 0,8298 0,7962 0,7379 0,6887 0,6266
0,99 0,9036 0,8913 0,8790 0,8590 0,8241 0,7674 0,7178 0,6546
1,00 0,9311 0,9204 0,9078 0,8872 0,8531 0,7962 0,7464 0,6823
1,01 0,9462 0,9462 0,9333 0,9162 0,8831 0,8241 0,7745 0,7096
1,02 0,9572 0,9661 0,9594 0,9419 0,9099 0,8531 0,8035 0,7379
1,05 0,9840 0,9954 1,0186 1,0162 0,9886 0,9354 0,8872 0,8222
1,10 1,0162 1,0280 1,0593 1,0990 1,1015 1,0617 1,0186 0,9572
1,15 1,0375 1,0520 1,0814 1,1376 1,1858 1,1722 1,1403 1,0864
1,20 1,0544 1,0691 1,0990 1,1588 1,2388 1,2647 1,2474 1,2050
1,30 1,0715 1,0914 1,1194 1,1776 1,2853 1,3868 1,4125 1,4061
1,40 1,0814 1,0990 1,1298 1,1858 1,2942 1,4488 1,5171 1,5524
1,50 1,0864 1,1041 1,1350 1,1858 1,2942 1,4689 1,5740 1,6520
1,60 1,0864 1,1041 1,1350 1,1858 1,2883 1,4689 1,5996 1,7140
1,70 1,0864 1,1041 1,1324 1,1803 1,2794 1,4622 1,6033 1,7458
1,80 1,0839 1,1015 1,1298 1,1749 1,2706 1,4488 1,5959 1,7620
1,90 1,0814 1,0990 1,1272 1,1695 1,2618 1,4355 1,5849 1,7620
2,00 1,0814 1,0965 1,1220 1,1641 1,2503 1,4191 1,5704 1,7539
2,20 1,0765 1,0914 1,1143 1,1535 1,2331 1,3900 1,5346 1,7219
2,40 1,0715 1,0864 1,1066 1,1429 1,2190 1,3614 1,4997 1,6866
2,60 1,0666 1,0814 1,1015 1,1350 1,2023 1,3397 1,4689 1,6482
2,80 1,0641 1,0765 1,0940 1,1272 1,1912 1,3183 1,4388 1,6144
3,00 1,0593 1,0715 1,0889 1,1194 1,1803 1,3002 1,4158 1,5813
3,50 1,0520 1,0617 1,0789 1,1041 1,1561 1,2618 1,3614 1,5101
4,00 1,0471 1,0544 1,0691 1,0914 1,1403 1,2303 1,3213 1,4555
You might also like
- Dispensa TermodinamicaDocument142 pagesDispensa TermodinamicaCamillo ForcellaNo ratings yet
- Meccanica Dei Fluidi - Lezioni Di FluidodinamicaDocument286 pagesMeccanica Dei Fluidi - Lezioni Di FluidodinamicaMauro MenchiniNo ratings yet
- Lezioni Di Fisica Tecnica - Termodinamica - Dassù InzoliDocument180 pagesLezioni Di Fisica Tecnica - Termodinamica - Dassù InzoliMarco De Donno100% (1)
- Dispensa TrasmissioneDocument98 pagesDispensa Trasmissioneantoniopavone2384100% (1)
- Appunti Di Analitica IIDocument45 pagesAppunti Di Analitica IImatteomonaiNo ratings yet
- Accumulatori per impianti ad energia rinnovabileFrom EverandAccumulatori per impianti ad energia rinnovabileRating: 2 out of 5 stars2/5 (1)
- Termodinamica Dell'ingegneria Chimica - Soluzioni EserciziDocument33 pagesTermodinamica Dell'ingegneria Chimica - Soluzioni EserciziMatteo TempestiNo ratings yet
- La relatività da Lorentz a Einstein.: Una guida per principianti, perplessi e scienziati sperimentali.From EverandLa relatività da Lorentz a Einstein.: Una guida per principianti, perplessi e scienziati sperimentali.No ratings yet
- Nomenclatura chimica inorganica. Reazioni redox. Principi di stechiometriaFrom EverandNomenclatura chimica inorganica. Reazioni redox. Principi di stechiometriaNo ratings yet
- Classificazione AcciaiDocument38 pagesClassificazione AcciaiLorenzo SantiNo ratings yet
- Esercizi Svolti Fisica TecnicaDocument74 pagesEsercizi Svolti Fisica TecnicaIlenia Raperonzolo GabrieleNo ratings yet
- Esercizi di fisica: termodinamica e trasmissione del caloreFrom EverandEsercizi di fisica: termodinamica e trasmissione del caloreRating: 5 out of 5 stars5/5 (1)
- Gestione di un progetto con Microsoft Project 2010 - AdvancedFrom EverandGestione di un progetto con Microsoft Project 2010 - AdvancedNo ratings yet
- Energy manager: Una professione vincente al servizio di imprese ed enti pubbliciFrom EverandEnergy manager: Una professione vincente al servizio di imprese ed enti pubbliciNo ratings yet
- Fenomeni Di Trasporto - Parte 1Document128 pagesFenomeni Di Trasporto - Parte 1Francesco TrevisanNo ratings yet
- Matematica vettoriale, matriciale e tensorialeFrom EverandMatematica vettoriale, matriciale e tensorialeRating: 5 out of 5 stars5/5 (1)
- Costruzione Di Macchine 2 - DispensaDocument168 pagesCostruzione Di Macchine 2 - DispensalucaverdeNo ratings yet
- Esercizi di matematica: successioni e serie di funzioniFrom EverandEsercizi di matematica: successioni e serie di funzioniRating: 5 out of 5 stars5/5 (1)
- Esercizi di matematica: equazioni integrali e integro-differenzialiFrom EverandEsercizi di matematica: equazioni integrali e integro-differenzialiNo ratings yet
- Esercizi IC1 9 CFUDocument57 pagesEsercizi IC1 9 CFUTutti Ganti A PezziNo ratings yet
- Materiali MettaliciDocument344 pagesMateriali Mettalicisankou7No ratings yet
- Reattori CataliticiDocument28 pagesReattori CataliticijeinerNo ratings yet
- Esercizi di fisica per licei: fluidodinamica e termodinamicaFrom EverandEsercizi di fisica per licei: fluidodinamica e termodinamicaRating: 5 out of 5 stars5/5 (1)
- Algebra Lineare e Geometria OttimoDocument235 pagesAlgebra Lineare e Geometria Ottimo20841No ratings yet
- Sivuchin - Fisica Atomica - (Corso Di Fisica Generale V Parte Prima) - Mir - Estere (1989)Document434 pagesSivuchin - Fisica Atomica - (Corso Di Fisica Generale V Parte Prima) - Mir - Estere (1989)Leonardo RennaNo ratings yet
- 0308 - Metallurgia E Saldabilità Degli Acciai Inossidabili - Istituto Italiano SaldaturaDocument48 pages0308 - Metallurgia E Saldabilità Degli Acciai Inossidabili - Istituto Italiano SaldaturaMariantoniettaSpallutoNo ratings yet
- SyngasDocument15 pagesSyngasLuca MarlettaNo ratings yet
- Climatizzazione di edifici con pompe di calore geotermiche. Analisi termodinamica ed economicaFrom EverandClimatizzazione di edifici con pompe di calore geotermiche. Analisi termodinamica ed economicaRating: 4 out of 5 stars4/5 (1)
- (Golia) Fluidodinamica (2005)Document240 pages(Golia) Fluidodinamica (2005)jonathan.gelli5153No ratings yet
- Controlli AutomaticiDocument188 pagesControlli AutomaticiivanNo ratings yet
- Turbine AssialiDocument44 pagesTurbine Assialiannalisa.d.orazio5738No ratings yet
- Esercizi di fisica per licei: cinematica, dinamica e staticaFrom EverandEsercizi di fisica per licei: cinematica, dinamica e staticaRating: 5 out of 5 stars5/5 (1)
- Chi MicaDocument574 pagesChi MicarodomontanoNo ratings yet
- Riassunti Di Scienza e Tecnologia Dei MaterialiDocument15 pagesRiassunti Di Scienza e Tecnologia Dei MaterialiGiovanniNo ratings yet
- Analisi Matematica 2Document313 pagesAnalisi Matematica 2Gian ClauNo ratings yet
- Temi Esame Fisica TecnicaDocument110 pagesTemi Esame Fisica Tecnicaomar_891422No ratings yet
- EserciziarioTA 2011Document73 pagesEserciziarioTA 2011GiuliadfNo ratings yet
- Dispense 2012-2013 Materiali MetalliciDocument261 pagesDispense 2012-2013 Materiali MetalliciAlessandroAbate0% (1)
- Impianti Turbine A VaporeDocument41 pagesImpianti Turbine A VaporeantoniomambroNo ratings yet
- Analisi 2 - GobbinoDocument250 pagesAnalisi 2 - GobbinoAndrea AnnunziataNo ratings yet
- Storia Dell'ammoniacaDocument8 pagesStoria Dell'ammoniacaBiagio CastronovoNo ratings yet
- Fisica TecnicaDocument84 pagesFisica TecnicaAndrea Di LorenzoNo ratings yet
- Dispensa Fisica TecnicaDocument465 pagesDispensa Fisica Tecnicayoelg1No ratings yet
- Azionamenti Dei Sistemi MeccaniciDocument127 pagesAzionamenti Dei Sistemi MeccaniciMassimo Moroni50% (2)
- Lezioni Di Scienza Delle Costruzioni, Claudio Franciosi 30 Giugno 2018 216pp PARTE IDocument216 pagesLezioni Di Scienza Delle Costruzioni, Claudio Franciosi 30 Giugno 2018 216pp PARTE IDjRacksNo ratings yet
- Lezione Facciorusso CorsoSiena 2feb012Document86 pagesLezione Facciorusso CorsoSiena 2feb012ingchiara85No ratings yet
- Fisica 24-25Document6 pagesFisica 24-25Davide StefaniNo ratings yet
- Estatica. Ingenieria MecanicaDocument1 pageEstatica. Ingenieria MecanicaW.L. PepeNo ratings yet
- 13 Moto Rettilineo Uniformemente Accelerato PDFDocument7 pages13 Moto Rettilineo Uniformemente Accelerato PDFAlessandro AlpiNo ratings yet
- 2419 Ese3Document2 pages2419 Ese3Sal MomiNo ratings yet
- Stampaggio LamieraDocument28 pagesStampaggio LamieraEnrico BoeriNo ratings yet
- Esercizi Parte2Document15 pagesEsercizi Parte2gian fratmNo ratings yet