You might also like
- OPRD-Hexanoyl Chloride PDFDocument6 pagesOPRD-Hexanoyl Chloride PDFsrimuruganNo ratings yet
- Heterogeneous Asymmetric Diels-Alder Reactions Using A Copper-Chiral Bis (Oxazoline) Complex Immobilized On Mesoporous SilicaDocument5 pagesHeterogeneous Asymmetric Diels-Alder Reactions Using A Copper-Chiral Bis (Oxazoline) Complex Immobilized On Mesoporous SilicaJC Jane BarnesNo ratings yet
- A-ring modifications on the triazafluorenone core structure provide potent mGluR1 antagonistsDocument4 pagesA-ring modifications on the triazafluorenone core structure provide potent mGluR1 antagonistsTuyenHHCNo ratings yet
- COM 08 11328asdfdsDocument9 pagesCOM 08 11328asdfdsVictor NgNo ratings yet
- A Practical Synthesis of (-) - Kainic AcidDocument11 pagesA Practical Synthesis of (-) - Kainic AcidNikhil SarpateNo ratings yet
- Total Synthesis of SwaisonineDocument7 pagesTotal Synthesis of SwaisonineVida Faith GalvezNo ratings yet
- A Stereocontrolled Synthetic Route To The C1 Pamamycin-607Document4 pagesA Stereocontrolled Synthetic Route To The C1 Pamamycin-607Devendar UradiNo ratings yet
- Basudeb Basu Et Al - A Simple Protocol For Direct Reductive Amination of Aldehydes and Ketones Using Potassium Formate and Catalytic Palladium AcetateDocument3 pagesBasudeb Basu Et Al - A Simple Protocol For Direct Reductive Amination of Aldehydes and Ketones Using Potassium Formate and Catalytic Palladium AcetateRoundSTICNo ratings yet
- DPS 2011 2 6 127 131Document5 pagesDPS 2011 2 6 127 131anuradha.d.bhat9860No ratings yet
- Bioorganic & Medicinal Chemistry (2001), 9 (10), 2693-2708Document16 pagesBioorganic & Medicinal Chemistry (2001), 9 (10), 2693-2708rrgodboleNo ratings yet
- Uses of 2-Ethoxy-4 (3H) Quinazolinone in Synthesis of Quinazoline and Quinazolinone Derivatives of Antimicrobial Activity: The Solvent EffectDocument12 pagesUses of 2-Ethoxy-4 (3H) Quinazolinone in Synthesis of Quinazoline and Quinazolinone Derivatives of Antimicrobial Activity: The Solvent Effectkhaliddarwish1962No ratings yet
- 1553 FTPDocument4 pages1553 FTPBedanta BorahNo ratings yet
- Organic Letters (2008), 10 (17), 3907-3909Document3 pagesOrganic Letters (2008), 10 (17), 3907-3909James TianNo ratings yet
- Apertura EpoxidoDocument6 pagesApertura EpoxidoLuis MartinezNo ratings yet
- R. Jason Herr - A Whirlwind Tour of Current Mitsunobu ChemistryDocument36 pagesR. Jason Herr - A Whirlwind Tour of Current Mitsunobu ChemistryRoundSTICNo ratings yet
- ChemMedChem (2009), 4 (8), 1269-1272Document4 pagesChemMedChem (2009), 4 (8), 1269-1272James TianNo ratings yet
- Gian Luca Araldi Et Al - An Enantioselective Synthesis of 2-Alkyl-3-Phenyltropanes by An Asymmetric 1,3-Dipolar Cycloaddition ReactionDocument2 pagesGian Luca Araldi Et Al - An Enantioselective Synthesis of 2-Alkyl-3-Phenyltropanes by An Asymmetric 1,3-Dipolar Cycloaddition ReactionGummyColaNo ratings yet
- P 19900000945Document10 pagesP 19900000945Yi-Yan TsaoNo ratings yet
- The Biocatalyzed Stereoselective Preparation of Polycyclic CyanohydrinsDocument7 pagesThe Biocatalyzed Stereoselective Preparation of Polycyclic CyanohydrinsDaigo G MamaniNo ratings yet
- The Chemistry of The Aminochromes: Part Vi. The Reaction O F Adrenochrome With Glutathione' "3Document7 pagesThe Chemistry of The Aminochromes: Part Vi. The Reaction O F Adrenochrome With Glutathione' "3Nstm3No ratings yet
- ExcessDocument6 pagesExcessAzbmNo ratings yet
- Can. J. Chem. 49, 1071-1084 (1971) - Methyl 4-BromobutyrateDocument15 pagesCan. J. Chem. 49, 1071-1084 (1971) - Methyl 4-Bromobutyratesunil_vaman_joshiNo ratings yet
- Development of A Stereoselective Practical Synthetic Route To Indolmycin, A Candidate Anti-H. Pylori AgentDocument5 pagesDevelopment of A Stereoselective Practical Synthetic Route To Indolmycin, A Candidate Anti-H. Pylori AgentMaria-LuizaGhidrasanNo ratings yet
- Umihara Et Al-2017-Chemistry - A European JournalDocument3 pagesUmihara Et Al-2017-Chemistry - A European JournalNathalia MojicaNo ratings yet
- Solvent Free Green Synthesis of 5-Arylidine Barbituric Acid Derivatives Catalyzed by Copper Oxide NanoparticlesDocument6 pagesSolvent Free Green Synthesis of 5-Arylidine Barbituric Acid Derivatives Catalyzed by Copper Oxide NanoparticlesbaskhemNo ratings yet
- DEMETHYLATION PROCEDUREDocument4 pagesDEMETHYLATION PROCEDUREFelipe MonteroNo ratings yet
- Leuckart ReactionDocument3 pagesLeuckart ReactionKybernetikum100% (1)
- Diastereoselective Synthesis of Homo-N, O-NucleosidesDocument8 pagesDiastereoselective Synthesis of Homo-N, O-Nucleosidesapi-19793040No ratings yet
- Poly EneDocument3 pagesPoly EneMohammed TarekNo ratings yet
- Glyconanoparticles Activity 3Document3 pagesGlyconanoparticles Activity 3ЛуизАпазаТ.No ratings yet
- Eur. J, 2010, 16, 6509-6517 Reek Anti-HalpernDocument9 pagesEur. J, 2010, 16, 6509-6517 Reek Anti-HalpernszbaloghNo ratings yet
- PNP Pourmousavi2017Document31 pagesPNP Pourmousavi2017sonNo ratings yet
- Annulation of Imidazolines With Bis-Electrophiles: Synthesis of Imidazo (1,2-A) PyridinesDocument10 pagesAnnulation of Imidazolines With Bis-Electrophiles: Synthesis of Imidazo (1,2-A) PyridinesboksabbNo ratings yet
- MMC 1Document6 pagesMMC 1umesh123patilNo ratings yet
- Angewandte Chemie, Vol. 46 Nb. 7 (2007) P. 1066 - 1070Document5 pagesAngewandte Chemie, Vol. 46 Nb. 7 (2007) P. 1066 - 1070rrgodboleNo ratings yet
- Synthesis of Seven-Membered 1,5-Anhydrocarbasugars and CycloheptanesDocument4 pagesSynthesis of Seven-Membered 1,5-Anhydrocarbasugars and CycloheptanesNGsalunkheNo ratings yet
- Synthesis of Using The Nanocomposite Catalyst: Biodiesel Mg/Al/Zn Hydrotalcite/SBA-15Document24 pagesSynthesis of Using The Nanocomposite Catalyst: Biodiesel Mg/Al/Zn Hydrotalcite/SBA-15Vikash ChandravanshiNo ratings yet
- Anthony R. Lingham Et Al - Studies Towards The Synthesis of Salvinorin ADocument9 pagesAnthony R. Lingham Et Al - Studies Towards The Synthesis of Salvinorin ABic0000No ratings yet
- Articulo Quimica Medicinal 3Document16 pagesArticulo Quimica Medicinal 3Jose Antonio Espinosa TorresNo ratings yet
- Despre MoleculeDocument14 pagesDespre MoleculeAlexa IordacheNo ratings yet
- CR 100258 KDocument35 pagesCR 100258 KzoyudgNo ratings yet
- A General Method For The Quaternization of N, N-Dimethyl Benzylamines With Long Chain N-AlkylbromidesDocument4 pagesA General Method For The Quaternization of N, N-Dimethyl Benzylamines With Long Chain N-AlkylbromidesirfanNo ratings yet
- Angew. Chem. Int. Ed. 2015, 54, 12134 - 12138Document5 pagesAngew. Chem. Int. Ed. 2015, 54, 12134 - 12138ludoNo ratings yet
- Preparation & Bio-Chemical Identification of Series Organic CompoundsDocument9 pagesPreparation & Bio-Chemical Identification of Series Organic CompoundschemistryjournalNo ratings yet
- Stereoselective Syntheses of Allylic Amines Through Reduction of 1-Azadiene IntermediatesDocument9 pagesStereoselective Syntheses of Allylic Amines Through Reduction of 1-Azadiene IntermediatesYas DelgaditoNo ratings yet
- Direct Synthesis of Nitriles From Aldehydes in Ionic LiquidsDocument3 pagesDirect Synthesis of Nitriles From Aldehydes in Ionic LiquidschidambaramrNo ratings yet
- Synthesis, Spectral Characterization and In-Vitro Analysis of Some Novel Title Chalcones DerivativesDocument5 pagesSynthesis, Spectral Characterization and In-Vitro Analysis of Some Novel Title Chalcones DerivativesSudhanshu Kumar JhaNo ratings yet
- Oleh: Kelompok 3 Budiman Yasir N012171029 Risna N012171012 Rismayanti Fauziah N012171022Document41 pagesOleh: Kelompok 3 Budiman Yasir N012171029 Risna N012171012 Rismayanti Fauziah N012171022yunitaNo ratings yet
- Changiz Karami, Keivan Ghodrati, Mina Izadi, Azita Farrokh, Sedigheh Jafari, Maryam Mahmoudiyani, and Nahid HaghnazariDocument4 pagesChangiz Karami, Keivan Ghodrati, Mina Izadi, Azita Farrokh, Sedigheh Jafari, Maryam Mahmoudiyani, and Nahid HaghnazariMiriam GarciaNo ratings yet
- JOC 1978 (43) 2320 - SasakiDocument6 pagesJOC 1978 (43) 2320 - SasakigioLXVNo ratings yet
- Molecules: Synthesis of Glycosides of Glucuronic, Galacturonic and Mannuronic Acids: An OverviewDocument36 pagesMolecules: Synthesis of Glycosides of Glucuronic, Galacturonic and Mannuronic Acids: An OverviewYuh HaurNo ratings yet
- Preparation and Pyrolysis of 1 - (Pyrazol-5-Yl) - 1,2,3-Triazoles and Related CompoundsDocument6 pagesPreparation and Pyrolysis of 1 - (Pyrazol-5-Yl) - 1,2,3-Triazoles and Related CompoundsChandra ReddyNo ratings yet
- Tetrahedron Letters: Shan Wang, Jian Wang, Rui Guo, Gao Wang, Shan-Yong Chen, Xiao-Qi YuDocument4 pagesTetrahedron Letters: Shan Wang, Jian Wang, Rui Guo, Gao Wang, Shan-Yong Chen, Xiao-Qi Yumarkbronson2009No ratings yet
- Substituted Pyrrolo (3,4-A) Carbazoles From Reactions Between 3 - (1-Methoxy-Vinyl) Indoles and MaleimidesDocument11 pagesSubstituted Pyrrolo (3,4-A) Carbazoles From Reactions Between 3 - (1-Methoxy-Vinyl) Indoles and MaleimidesShubham RajputNo ratings yet
- 1 s2.0 S0020169305006274 Main PDFDocument5 pages1 s2.0 S0020169305006274 Main PDFhenry martinez quiñonezNo ratings yet
- Manuscript DraftDocument20 pagesManuscript DraftsadaNo ratings yet
- Synthesis of Halogenated 1H-Cyclohepta (2,1-b:3,4-b') Diindoles and Their Nucleophilic Aromatic Substitution ReactionsDocument9 pagesSynthesis of Halogenated 1H-Cyclohepta (2,1-b:3,4-b') Diindoles and Their Nucleophilic Aromatic Substitution Reactionstuấn anhNo ratings yet
- Sustainable synthesis of ciclopentene derivatives through multicomponent reactions in continuous flow regimeFrom EverandSustainable synthesis of ciclopentene derivatives through multicomponent reactions in continuous flow regimeNo ratings yet
- Transition Metal-Catalyzed Pyridine Synthesis: Transition Metal-Catalyzed Heterocycle Synthesis SeriesFrom EverandTransition Metal-Catalyzed Pyridine Synthesis: Transition Metal-Catalyzed Heterocycle Synthesis SeriesNo ratings yet
- Carbohydrate Chemistry—8: Plenary Lectures Presented at the Eighth International Symposium on Carbohydrate Chemistry, Kyoto, Japan 16 - 20 August 1976From EverandCarbohydrate Chemistry—8: Plenary Lectures Presented at the Eighth International Symposium on Carbohydrate Chemistry, Kyoto, Japan 16 - 20 August 1976K. OnoderaNo ratings yet
- Effect of Fiber Length on Mechanical Properties of Coir Fiber Reinforced Epoxy CompositesDocument6 pagesEffect of Fiber Length on Mechanical Properties of Coir Fiber Reinforced Epoxy Compositesbrar_harmanpreet01No ratings yet
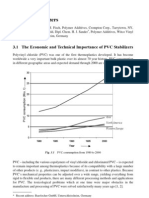
- PVC StabilisersDocument20 pagesPVC StabilisersGustavo SalazarNo ratings yet
- Cmos Process FlowDocument29 pagesCmos Process FlowPushparaj Karu100% (1)
- Certificate of AnalysisDocument1 pageCertificate of AnalysisMiguel CruzNo ratings yet
- 0271.SAS-BR-Inorganics Siral Siralox WEBDocument16 pages0271.SAS-BR-Inorganics Siral Siralox WEBMrutunjay PatraNo ratings yet
- PharmaDocument147 pagesPharmaDrChauhanNo ratings yet
- OIL RS ULTRA MSDS EngDocument10 pagesOIL RS ULTRA MSDS Engmohamed.elattar1995No ratings yet
- BykDocument5 pagesByksamratsamudraguptaNo ratings yet
- Design of Process EquipmentDocument175 pagesDesign of Process EquipmentAravind MadhuNo ratings yet
- Chap. 4.1Document11 pagesChap. 4.1Azizah UlfaNo ratings yet
- Wastewater Treatment OverviewDocument55 pagesWastewater Treatment OverviewShorOuq Mohammed Malkawi100% (1)
- Easy Soap Making RecipesDocument38 pagesEasy Soap Making Recipesgeorge kamauNo ratings yet
- Experiment 4 CODDocument3 pagesExperiment 4 CODNurul Noorfazleen78% (9)
- HPL Additives Training ReportDocument34 pagesHPL Additives Training ReportAjay Shekhawat100% (1)
- SiP ExeDocument8 pagesSiP ExeJomari Villadelrey100% (1)
- Types Compressors Used in HVACDocument12 pagesTypes Compressors Used in HVACQasimIbrarNo ratings yet
- Microbiological Quality of Different Brands of Talcum Powder in IndiaDocument6 pagesMicrobiological Quality of Different Brands of Talcum Powder in IndiaJohan SukweenadhiNo ratings yet
- Baqueta2019 JFS SensorialDocument10 pagesBaqueta2019 JFS SensorialTanChantreaNo ratings yet
- Kevlar Composite JurnalDocument10 pagesKevlar Composite JurnalJakbrother 28No ratings yet
- P5 Conjugated DienesDocument52 pagesP5 Conjugated DienesShirl Angelee OcampoNo ratings yet
- Type C: Banded Elastomeric BearingsDocument4 pagesType C: Banded Elastomeric Bearingsflorin_iacob2001No ratings yet
- Chemical KineticsDocument52 pagesChemical KineticsdhananjaylandgeNo ratings yet
- Integrative Oncology GuideDocument26 pagesIntegrative Oncology GuideSWAPNIL DWIVEDINo ratings yet
- Diagrama de FlujoDocument18 pagesDiagrama de FlujoLAURA KATHERINE CENTENO JAIMESNo ratings yet
- Horn Fly and Stable FlyDocument39 pagesHorn Fly and Stable Flylady ann jimenezNo ratings yet
- Week 4 - ReadingDocument2 pagesWeek 4 - Readingapi-254428474100% (1)
- Fosroc Conbextra GP2Document4 pagesFosroc Conbextra GP2awsdhnjkklmNo ratings yet
- SPEC Tamanu OilDocument1 pageSPEC Tamanu OilSuci Desriana RSNo ratings yet
- Dimesiones Tee's EnvolventeDocument4 pagesDimesiones Tee's EnvolventeJuan Carlos Dominguez Bta0% (1)
- 1 Bio ChemDocument23 pages1 Bio ChemRuwi Loren Villocino100% (2)