You might also like
- The Islamic Perspective On Stem Cell ResearchDocument18 pagesThe Islamic Perspective On Stem Cell ResearchMutia LailaniNo ratings yet
- Nasopharyngeal Ca DR - JackyDocument18 pagesNasopharyngeal Ca DR - JackyMutia LailaniNo ratings yet
- Recommendations of Euthanasia For Experimental Animals PDFDocument24 pagesRecommendations of Euthanasia For Experimental Animals PDFMutia LailaniNo ratings yet
- Lani DAFTAR PUSTAKADocument2 pagesLani DAFTAR PUSTAKAMutia LailaniNo ratings yet
- Rheumatic Chorea in Northern Australia: A Clinical and Epidemiological StudyDocument6 pagesRheumatic Chorea in Northern Australia: A Clinical and Epidemiological StudyMutia LailaniNo ratings yet
- Prospective Comparison of Clinical and Echocardiographic Diagnosis of Rheumatic Carditis: Long Term Follow Up of Patients With Subclinical DiseaseDocument4 pagesProspective Comparison of Clinical and Echocardiographic Diagnosis of Rheumatic Carditis: Long Term Follow Up of Patients With Subclinical DiseaseMutia LailaniNo ratings yet
- BST - Skizoafektif Tipe ManikDocument16 pagesBST - Skizoafektif Tipe ManikMutia LailaniNo ratings yet
- Pessary InsertionDocument2 pagesPessary InsertionMutia LailaniNo ratings yet
- Parturition Events and Risk of Urinary Incontinence in Later LifeDocument16 pagesParturition Events and Risk of Urinary Incontinence in Later LifeMutia LailaniNo ratings yet
- Clinical Laboratories: Point of Care TestingDocument3 pagesClinical Laboratories: Point of Care TestingMutia LailaniNo ratings yet
- Shoe Dog: A Memoir by the Creator of NikeFrom EverandShoe Dog: A Memoir by the Creator of NikeRating: 4.5 out of 5 stars4.5/5 (537)
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeFrom EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeRating: 4 out of 5 stars4/5 (5794)
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceFrom EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceRating: 4 out of 5 stars4/5 (895)
- The Yellow House: A Memoir (2019 National Book Award Winner)From EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Rating: 4 out of 5 stars4/5 (98)
- Grit: The Power of Passion and PerseveranceFrom EverandGrit: The Power of Passion and PerseveranceRating: 4 out of 5 stars4/5 (588)
- The Little Book of Hygge: Danish Secrets to Happy LivingFrom EverandThe Little Book of Hygge: Danish Secrets to Happy LivingRating: 3.5 out of 5 stars3.5/5 (400)
- The Emperor of All Maladies: A Biography of CancerFrom EverandThe Emperor of All Maladies: A Biography of CancerRating: 4.5 out of 5 stars4.5/5 (271)
- Never Split the Difference: Negotiating As If Your Life Depended On ItFrom EverandNever Split the Difference: Negotiating As If Your Life Depended On ItRating: 4.5 out of 5 stars4.5/5 (838)
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyFrom EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyRating: 3.5 out of 5 stars3.5/5 (2259)
- On Fire: The (Burning) Case for a Green New DealFrom EverandOn Fire: The (Burning) Case for a Green New DealRating: 4 out of 5 stars4/5 (74)
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureFrom EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureRating: 4.5 out of 5 stars4.5/5 (474)
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryFrom EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryRating: 3.5 out of 5 stars3.5/5 (231)
- Team of Rivals: The Political Genius of Abraham LincolnFrom EverandTeam of Rivals: The Political Genius of Abraham LincolnRating: 4.5 out of 5 stars4.5/5 (234)
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaFrom EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaRating: 4.5 out of 5 stars4.5/5 (266)
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersFrom EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersRating: 4.5 out of 5 stars4.5/5 (345)
- The Unwinding: An Inner History of the New AmericaFrom EverandThe Unwinding: An Inner History of the New AmericaRating: 4 out of 5 stars4/5 (45)
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreFrom EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreRating: 4 out of 5 stars4/5 (1090)
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)From EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Rating: 4.5 out of 5 stars4.5/5 (121)
- Her Body and Other Parties: StoriesFrom EverandHer Body and Other Parties: StoriesRating: 4 out of 5 stars4/5 (821)
- 8 13 PDFDocument7 pages8 13 PDFVillycia LNo ratings yet
- Matrices Progresivas Escala Coloreada Manual PDFDocument27 pagesMatrices Progresivas Escala Coloreada Manual PDFMaría LuzNo ratings yet
- Parts of TheEyesDocument4 pagesParts of TheEyesAden Keith Paus ValdezNo ratings yet
- PCR Primer Design PDFDocument62 pagesPCR Primer Design PDFnesariaNo ratings yet
- BIO MCQS by IlmiDocument67 pagesBIO MCQS by IlmiZee waqarNo ratings yet
- Instructions Template For Freshman ResumeDocument2 pagesInstructions Template For Freshman ResumeGuillaumeINo ratings yet
- Mob Ot 31753002739420Document276 pagesMob Ot 31753002739420fabriziozaraNo ratings yet
- PG I IflashDocument4 pagesPG I IflashNIGHT tubeNo ratings yet
- 9780702072987-Book ChapterDocument2 pages9780702072987-Book ChaptervisiniNo ratings yet
- ScienceDocument7 pagesScienceDhwani ChitrodaNo ratings yet
- Reading Genre Descriptive-Text PDFDocument2 pagesReading Genre Descriptive-Text PDFEro TradingNo ratings yet
- Q4 Week 3 4 GenBio2 EditedDocument12 pagesQ4 Week 3 4 GenBio2 EditedXyreen GalicinaoNo ratings yet
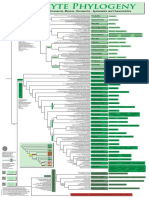
- Filogenia de Briofitos SensulatoDocument1 pageFilogenia de Briofitos SensulatoYuyitoS2714No ratings yet
- Colloidal Silver and Viral InfectionsDocument13 pagesColloidal Silver and Viral Infectionsspencerjonesy100% (7)
- Nano Dox CarrierDocument7 pagesNano Dox CarrierMuanfan SuwanNo ratings yet
- Plant Growth 2Document2 pagesPlant Growth 2Asnani AsudinNo ratings yet
- Daily Lesson Plan: 7e's ApproachDocument6 pagesDaily Lesson Plan: 7e's ApproachSonny MagdadaroNo ratings yet
- 1,4 ButanediolDocument6 pages1,4 ButanediolVictor VikeneNo ratings yet
- What Are Hormones?Document4 pagesWhat Are Hormones?Uzair SoomroNo ratings yet
- Nursing ProcessDocument23 pagesNursing Processjulesubayubay5428100% (6)
- Encyclopedia ES HealthDocument9 pagesEncyclopedia ES HealthGantsooj BNo ratings yet
- Hellagrolip Nitrogenous Brochure enDocument2 pagesHellagrolip Nitrogenous Brochure enBashir A. SialNo ratings yet
- AdatinaDocument1 pageAdatinaCRISTIELI IZABEL FOGUESATTONo ratings yet
- Apostolou Wang 2020Document11 pagesApostolou Wang 2020Riotgryph LepitorusNo ratings yet
- 1 CoverDocument8 pages1 CoverMika Anjelia SebayangNo ratings yet
- Diffusion and Osmosis Worksheet: Garrison O'Level Campus Cantt, QuettaDocument6 pagesDiffusion and Osmosis Worksheet: Garrison O'Level Campus Cantt, QuettagulminaNo ratings yet
- Non MedelianDocument29 pagesNon MedelianKlaudette Collin PaynorNo ratings yet
- Pronunciation Stress Language ExchangesDocument5 pagesPronunciation Stress Language ExchangesHàNhậtNguyễnNo ratings yet
- Een House Monitoring and Spray Water SystemDocument2 pagesEen House Monitoring and Spray Water SystemDinesh KumarNo ratings yet
- Duchenne Muscular DystrophyDocument32 pagesDuchenne Muscular DystrophyMr.P.Ramesh, Faculty of Physiotherapy, SRUNo ratings yet