You might also like
- Hdout Chap 10 5210Document62 pagesHdout Chap 10 5210salinips3No ratings yet
- Solved Examples - Solutions, Class 12, Chemistry - EduRevDocument24 pagesSolved Examples - Solutions, Class 12, Chemistry - EduRevsalinips333% (3)
- 11Document40 pages11salinips3No ratings yet
- MSC Org Chem Notes GDDocument256 pagesMSC Org Chem Notes GDsalinips350% (2)
- Prepared by José Laboy, MS: Chem 343 - Organic Reactions Chapter 5 & 7Document1 pagePrepared by José Laboy, MS: Chem 343 - Organic Reactions Chapter 5 & 7salinips3No ratings yet
- Spectroscopy of Organic CompoundsDocument36 pagesSpectroscopy of Organic Compoundsnandhini_lgc0% (1)
- B. An Introduction To (Bio) Inorganic Chemistry: The Metal-Ligand InteractionDocument9 pagesB. An Introduction To (Bio) Inorganic Chemistry: The Metal-Ligand Interactionsalinips3No ratings yet
- Lecture 31Document8 pagesLecture 31Miguel RochaNo ratings yet
- Nuclear ChemistryDocument9 pagesNuclear Chemistrysalinips3No ratings yet
- The Metallic BondDocument20 pagesThe Metallic BondhumejiasNo ratings yet
- Nuclear ChemistryDocument9 pagesNuclear Chemistrysalinips3No ratings yet
- Molecular TermsDocument3 pagesMolecular Termssalinips3No ratings yet
- 2 HemocyaninDocument37 pages2 Hemocyaninsalinips3No ratings yet
- Inorganic Chemistry: Co and Nahco Co CR O SoDocument2 pagesInorganic Chemistry: Co and Nahco Co CR O Sosalinips3No ratings yet
- Part FN Worked Examples PDFDocument5 pagesPart FN Worked Examples PDFtizazu dfeteneNo ratings yet
- PChemII Lecture 07 SpectroscopyII P2Document17 pagesPChemII Lecture 07 SpectroscopyII P2salinips3No ratings yet
- Chemistry Experimental Chemistry PDFDocument31 pagesChemistry Experimental Chemistry PDFSiddharth RatnamNo ratings yet
- CDDocument6 pagesCDsalinips3No ratings yet
- Molecular Rotational SpectrosDocument41 pagesMolecular Rotational SpectrosAmar RoyNo ratings yet
- TaylorDocument5 pagesTaylorsalinips3No ratings yet
- Organic PhotovoltaicsDocument3 pagesOrganic Photovoltaicssalinips3No ratings yet
- Nanorib Hair ChipsDocument2 pagesNanorib Hair Chipssalinips3No ratings yet
- Steps in Crystal Structure Analysis A. Growing Crystals B. Microscopic ExaminationDocument1 pageSteps in Crystal Structure Analysis A. Growing Crystals B. Microscopic Examinationsalinips3No ratings yet
- Stereo ChemDocument37 pagesStereo Chemsalinips3No ratings yet
- Organic ChemistryDocument57 pagesOrganic ChemistryKunjal100% (1)
- FluorescenceDocument12 pagesFluorescencesalinips3No ratings yet
- Mass SpectrosDocument111 pagesMass SpectrosMohammed75% (8)
- Organic ChemistryDocument57 pagesOrganic ChemistryKunjal100% (1)
- Higher Algebra - Hall & KnightDocument593 pagesHigher Algebra - Hall & KnightRam Gollamudi100% (2)
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeFrom EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeRating: 4 out of 5 stars4/5 (5794)
- The Yellow House: A Memoir (2019 National Book Award Winner)From EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Rating: 4 out of 5 stars4/5 (98)
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryFrom EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryRating: 3.5 out of 5 stars3.5/5 (231)
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceFrom EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceRating: 4 out of 5 stars4/5 (895)
- The Little Book of Hygge: Danish Secrets to Happy LivingFrom EverandThe Little Book of Hygge: Danish Secrets to Happy LivingRating: 3.5 out of 5 stars3.5/5 (400)
- Shoe Dog: A Memoir by the Creator of NikeFrom EverandShoe Dog: A Memoir by the Creator of NikeRating: 4.5 out of 5 stars4.5/5 (537)
- Never Split the Difference: Negotiating As If Your Life Depended On ItFrom EverandNever Split the Difference: Negotiating As If Your Life Depended On ItRating: 4.5 out of 5 stars4.5/5 (838)
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureFrom EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureRating: 4.5 out of 5 stars4.5/5 (474)
- Grit: The Power of Passion and PerseveranceFrom EverandGrit: The Power of Passion and PerseveranceRating: 4 out of 5 stars4/5 (588)
- The Emperor of All Maladies: A Biography of CancerFrom EverandThe Emperor of All Maladies: A Biography of CancerRating: 4.5 out of 5 stars4.5/5 (271)
- On Fire: The (Burning) Case for a Green New DealFrom EverandOn Fire: The (Burning) Case for a Green New DealRating: 4 out of 5 stars4/5 (74)
- Team of Rivals: The Political Genius of Abraham LincolnFrom EverandTeam of Rivals: The Political Genius of Abraham LincolnRating: 4.5 out of 5 stars4.5/5 (234)
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaFrom EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaRating: 4.5 out of 5 stars4.5/5 (266)
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersFrom EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersRating: 4.5 out of 5 stars4.5/5 (344)
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyFrom EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyRating: 3.5 out of 5 stars3.5/5 (2259)
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreFrom EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreRating: 4 out of 5 stars4/5 (1090)
- The Unwinding: An Inner History of the New AmericaFrom EverandThe Unwinding: An Inner History of the New AmericaRating: 4 out of 5 stars4/5 (45)
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)From EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Rating: 4.5 out of 5 stars4.5/5 (121)
- Her Body and Other Parties: StoriesFrom EverandHer Body and Other Parties: StoriesRating: 4 out of 5 stars4/5 (821)
- Who Has Allergies & Why: Allergies, Also Known As Allergic Diseases, Are A Number of Conditions Caused byDocument6 pagesWho Has Allergies & Why: Allergies, Also Known As Allergic Diseases, Are A Number of Conditions Caused byJun Dl CrzNo ratings yet
- Fire AlarmDocument18 pagesFire AlarmgauriNo ratings yet
- 1Document3 pages1Pradeep PunterNo ratings yet
- Barge 180Ft Deck Load Capacity & Strength-Rev1Document52 pagesBarge 180Ft Deck Load Capacity & Strength-Rev1Wahyu Codyr86% (7)
- Coulomb's Law and Electric Field Intensity: Engineering ElectromagneticsDocument24 pagesCoulomb's Law and Electric Field Intensity: Engineering ElectromagneticsKenn SenadosNo ratings yet
- Unit 21Document22 pagesUnit 21Yuni IndahNo ratings yet
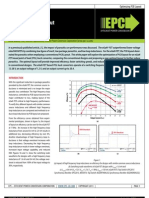
- Optimizing With eGaN FETsDocument6 pagesOptimizing With eGaN FETskhsniperNo ratings yet
- 9) Expt No - 9 (Halleffect)Document16 pages9) Expt No - 9 (Halleffect)Pollack Prittam ChoudhuryNo ratings yet
- Brunei 2Document16 pagesBrunei 2Eva PurnamasariNo ratings yet
- Las Mapeh 9 q2 w6 HealthDocument8 pagesLas Mapeh 9 q2 w6 HealthJemalyn Hibaya Lasaca100% (1)
- CSR and Sustainability Initiatives of Starbucks IncDocument20 pagesCSR and Sustainability Initiatives of Starbucks IncVidyut BanerjeeNo ratings yet
- Taiwan API Manufacturer ListDocument4 pagesTaiwan API Manufacturer Listkalyani dynamicsNo ratings yet
- Dobdsm 304Document39 pagesDobdsm 304LuisangelDueñasNo ratings yet
- Anaerobic Degradation of Palm Oil Mill Ef Uent (POME)Document8 pagesAnaerobic Degradation of Palm Oil Mill Ef Uent (POME)HusainiNo ratings yet
- 96-09302-00-01 Reva Technical Manual Inogen One G5Document18 pages96-09302-00-01 Reva Technical Manual Inogen One G5Paula Andrea MarulandaNo ratings yet
- Easy Guide For Fujitsu T901 LaptopDocument141 pagesEasy Guide For Fujitsu T901 LaptopElaineNo ratings yet
- Introduction To Food Analysis2020Document2 pagesIntroduction To Food Analysis2020Ĝĭdęŷ KîřöşNo ratings yet
- Draf Model LC 2024 Non TransferableDocument3 pagesDraf Model LC 2024 Non TransferablepresidenciaNo ratings yet
- Buk Uuuuuu UuuuuuuDocument92 pagesBuk Uuuuuu UuuuuuuJanaliyaNo ratings yet
- CEBUANO ERNESTO CODINA (Astonaut Hardware Designer)Document1 pageCEBUANO ERNESTO CODINA (Astonaut Hardware Designer)Dessirea FurigayNo ratings yet
- Philippine Airlines Reservation New Timings Dep - 230314 - 193643Document7 pagesPhilippine Airlines Reservation New Timings Dep - 230314 - 193643sophia buiserNo ratings yet
- BNB SB0114Document4 pagesBNB SB0114graziana100% (2)
- Basic Resistance Training GP5Document20 pagesBasic Resistance Training GP5matt.tubieron23No ratings yet
- Quick Start Guide For The Remote Access Dial-In Multiport Ethernet ModemDocument16 pagesQuick Start Guide For The Remote Access Dial-In Multiport Ethernet ModemdilipNo ratings yet
- Pathoftherosesyllabus2015 1 PDFDocument12 pagesPathoftherosesyllabus2015 1 PDFsperm100% (7)
- KIN-CN-STU-NW-0001 Puerto Real Towing Study of Kincardine 04Document44 pagesKIN-CN-STU-NW-0001 Puerto Real Towing Study of Kincardine 04RUBEN BARTOLOME GARCIA100% (1)
- Whirlpool Adg 789Document22 pagesWhirlpool Adg 789Laurentiu GramaNo ratings yet
- IJHIM 6 - Nur Husnina (36 SD 42)Document7 pagesIJHIM 6 - Nur Husnina (36 SD 42)RSU Sayang BundaNo ratings yet
- Factorisation PDFDocument3 pagesFactorisation PDFRaj Kumar0% (1)
- Government Schemes: Ministry of Agriculture and Farmers' WelfareDocument29 pagesGovernment Schemes: Ministry of Agriculture and Farmers' WelfareDushyant MudgalNo ratings yet