You might also like
- Shoe Dog: A Memoir by the Creator of NikeFrom EverandShoe Dog: A Memoir by the Creator of NikeRating: 4.5 out of 5 stars4.5/5 (537)
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeFrom EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeRating: 4 out of 5 stars4/5 (5794)
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceFrom EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceRating: 4 out of 5 stars4/5 (895)
- The Yellow House: A Memoir (2019 National Book Award Winner)From EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Rating: 4 out of 5 stars4/5 (98)
- Grit: The Power of Passion and PerseveranceFrom EverandGrit: The Power of Passion and PerseveranceRating: 4 out of 5 stars4/5 (588)
- The Little Book of Hygge: Danish Secrets to Happy LivingFrom EverandThe Little Book of Hygge: Danish Secrets to Happy LivingRating: 3.5 out of 5 stars3.5/5 (400)
- The Emperor of All Maladies: A Biography of CancerFrom EverandThe Emperor of All Maladies: A Biography of CancerRating: 4.5 out of 5 stars4.5/5 (271)
- Never Split the Difference: Negotiating As If Your Life Depended On ItFrom EverandNever Split the Difference: Negotiating As If Your Life Depended On ItRating: 4.5 out of 5 stars4.5/5 (838)
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyFrom EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyRating: 3.5 out of 5 stars3.5/5 (2259)
- On Fire: The (Burning) Case for a Green New DealFrom EverandOn Fire: The (Burning) Case for a Green New DealRating: 4 out of 5 stars4/5 (74)
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureFrom EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureRating: 4.5 out of 5 stars4.5/5 (474)
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryFrom EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryRating: 3.5 out of 5 stars3.5/5 (231)
- Team of Rivals: The Political Genius of Abraham LincolnFrom EverandTeam of Rivals: The Political Genius of Abraham LincolnRating: 4.5 out of 5 stars4.5/5 (234)
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaFrom EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaRating: 4.5 out of 5 stars4.5/5 (266)
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersFrom EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersRating: 4.5 out of 5 stars4.5/5 (345)
- The Unwinding: An Inner History of the New AmericaFrom EverandThe Unwinding: An Inner History of the New AmericaRating: 4 out of 5 stars4/5 (45)
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreFrom EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreRating: 4 out of 5 stars4/5 (1090)
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)From EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Rating: 4.5 out of 5 stars4.5/5 (121)
- Her Body and Other Parties: StoriesFrom EverandHer Body and Other Parties: StoriesRating: 4 out of 5 stars4/5 (821)
- TOK Assessed Student WorkDocument10 pagesTOK Assessed Student WorkPeter Jun Park100% (1)
- Finite State MachineDocument75 pagesFinite State Machinecall_asitNo ratings yet
- BIM and Big Data For Construction Cost ManagementDocument46 pagesBIM and Big Data For Construction Cost Managementlu09100% (1)
- Operation ManagementDocument4 pagesOperation ManagementHananiya GizawNo ratings yet
- 3D Tetris Cake Evening 2Document13 pages3D Tetris Cake Evening 2Subham KarmakarNo ratings yet
- Change LogDocument145 pagesChange LogelhohitoNo ratings yet
- End Points SubrogadosDocument3 pagesEnd Points SubrogadosAgustina AndradeNo ratings yet
- BS en 50216-6 2002Document18 pagesBS en 50216-6 2002Jeff Anderson Collins100% (3)
- Re BuyerDocument20 pagesRe BuyerElias OjuokNo ratings yet
- Blackstone The Dash Model #1610 Owner's ManualDocument53 pagesBlackstone The Dash Model #1610 Owner's ManualSydney Adam SteeleNo ratings yet
- Datasheet - HK ml7012-04 3364913Document22 pagesDatasheet - HK ml7012-04 3364913kami samaNo ratings yet
- PERSONAL DEVELOPMENT (What Is Personal Development?)Document37 pagesPERSONAL DEVELOPMENT (What Is Personal Development?)Ronafe Roncal GibaNo ratings yet
- BIO122 - CHAPTER 7 Part 1Document53 pagesBIO122 - CHAPTER 7 Part 1lili100% (1)
- Ebook Computer Forensics Principles and Practices 1St Edition Volonino Test Bank Full Chapter PDFDocument29 pagesEbook Computer Forensics Principles and Practices 1St Edition Volonino Test Bank Full Chapter PDFmundifycoucheefnhgl100% (10)
- Chapter 2.2 Quantitative Analysis NewDocument44 pagesChapter 2.2 Quantitative Analysis NewMinase TilayeNo ratings yet
- Ugtt April May 2019 NewDocument48 pagesUgtt April May 2019 NewSuhas SNo ratings yet
- Normal Consistency of Hydraulic CementDocument15 pagesNormal Consistency of Hydraulic CementApril Lyn SantosNo ratings yet
- Week 2 - Sulphur DyesDocument5 pagesWeek 2 - Sulphur DyesRR TNo ratings yet
- Assessment PN1096617Document14 pagesAssessment PN1096617Amr TarekNo ratings yet
- Gol GumbazDocument6 pagesGol Gumbazmnv_iitbNo ratings yet
- MIDTERM Exam - Programming 2 - 2SEM 2020Document3 pagesMIDTERM Exam - Programming 2 - 2SEM 2020Bab bidiNo ratings yet
- M4110 Leakage Reactance InterfaceDocument2 pagesM4110 Leakage Reactance InterfaceGuru MishraNo ratings yet
- TakeawaysDocument2 pagesTakeawaysapi-509552154No ratings yet
- Beginning Cosmetic ChemistryDocument1 pageBeginning Cosmetic ChemistrySergio Rugerio0% (1)
- CV TitchievDocument3 pagesCV TitchievIna FarcosNo ratings yet
- EUCLIDDocument3 pagesEUCLIDNandini MouryaNo ratings yet
- Terasaki FDP 2013Document40 pagesTerasaki FDP 2013MannyBaldonadoDeJesus100% (1)
- TAPPI T 810 Om-06 Bursting Strength of Corrugated and Solid FiberboardDocument5 pagesTAPPI T 810 Om-06 Bursting Strength of Corrugated and Solid FiberboardNguyenSongHaoNo ratings yet
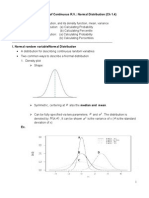
- Distribution of Continuous R.V.: Normal Distribution (CH 1.4) TopicsDocument7 pagesDistribution of Continuous R.V.: Normal Distribution (CH 1.4) TopicsPhạm Ngọc HòaNo ratings yet
- CNNPX310R-6P: General SpecificationsDocument5 pagesCNNPX310R-6P: General SpecificationsZoheir KacimiNo ratings yet