You might also like
- Non-Sickle HB Variants.091611Document44 pagesNon-Sickle HB Variants.091611Sumihar PasaribuNo ratings yet
- 75 PDFDocument4 pages75 PDFJR VendeventerNo ratings yet
- Automated Capillary Electrophoresis in The ScreeningDocument9 pagesAutomated Capillary Electrophoresis in The Screeningsomething privateNo ratings yet
- HPLC ThesisDocument21 pagesHPLC ThesisDolly SurelaNo ratings yet
- Peroxidase Activity of Hemoglobin - Haptoglobin ComplexesDocument14 pagesPeroxidase Activity of Hemoglobin - Haptoglobin ComplexesJoanne MachadoNo ratings yet
- Hemoglobin Called Hemoglobin S or Hbs or Sickle Hemoglobin, in The Red Blood CellsDocument13 pagesHemoglobin Called Hemoglobin S or Hbs or Sickle Hemoglobin, in The Red Blood CellsPhương Ly LêNo ratings yet
- HB Q-ThailandDocument6 pagesHB Q-Thailandladydew83No ratings yet
- The Thalassemias and Hemolytic AnemiasDocument43 pagesThe Thalassemias and Hemolytic AnemiasAnonymous elq7jZiSNo ratings yet
- Case Study: Rare Combination of α-Thalassemia Mutations and Sickle Cell DiseaseDocument5 pagesCase Study: Rare Combination of α-Thalassemia Mutations and Sickle Cell DiseaseFirdaus Al HafizNo ratings yet
- HbE-_THALASSEMIA_A_CASE_REPORTDocument2 pagesHbE-_THALASSEMIA_A_CASE_REPORTHarshika KDGNo ratings yet
- HemoglobinopathiesDocument51 pagesHemoglobinopathiesmontey007No ratings yet
- 6, Hemolytic AnemiaDocument42 pages6, Hemolytic AnemiaNouman HameedNo ratings yet
- 543Document3 pages543Mai MostafaNo ratings yet
- A Model for Gene Therapy: Gene Replacement in the Treatment of Sickle Cell Anemia and ThalassemiaFrom EverandA Model for Gene Therapy: Gene Replacement in the Treatment of Sickle Cell Anemia and ThalassemiaNo ratings yet
- HemoglobinopathiesDocument3 pagesHemoglobinopathiesMohamed Abdullahi OsmanNo ratings yet
- Alpha ThalassemiaDocument3 pagesAlpha ThalassemiaSravya PangaNo ratings yet
- Talasemia Hb-PatiDocument68 pagesTalasemia Hb-PatiNoer HafniNo ratings yet
- Blood 8Document7 pagesBlood 8ashokNo ratings yet
- HPLC PPT - KarishmaDocument76 pagesHPLC PPT - KarishmaDivya GauravNo ratings yet
- LORESCA - BB Ratio Activity 2Document14 pagesLORESCA - BB Ratio Activity 2Kaycee Gretz LorescaNo ratings yet
- Sickle Cell DiseaseDocument18 pagesSickle Cell DiseasedrheayNo ratings yet
- LECTURE 6 The ABO Blood Group SystemDocument64 pagesLECTURE 6 The ABO Blood Group SystemKrisyah Niqoule Valdez100% (2)
- Complete Genetics Disease ChartDocument14 pagesComplete Genetics Disease ChartJames FlanneryNo ratings yet
- J. Biol. Chem.-2016-Bissé-jbc.M116.764274Document28 pagesJ. Biol. Chem.-2016-Bissé-jbc.M116.764274Patricia FernandesNo ratings yet
- Malik2005 PDFDocument12 pagesMalik2005 PDFYul VulturiNo ratings yet
- 1303 Manuscript 5599 1 10 20170901Document8 pages1303 Manuscript 5599 1 10 20170901hariprem26No ratings yet
- THALASSEMIADocument30 pagesTHALASSEMIAjismi vallachiraNo ratings yet
- 3-T and Sicke Cell Disease 2016Document68 pages3-T and Sicke Cell Disease 2016ThaveeshaLindsayWhiteNo ratings yet
- IJTK 17 (3) 598-601 Thalasmia2Document4 pagesIJTK 17 (3) 598-601 Thalasmia2Jatin AnandNo ratings yet
- Blood Group Types and MethodsDocument64 pagesBlood Group Types and MethodsDranreb Berylle MasangkayNo ratings yet
- Haptoglobin - PubMedDocument1 pageHaptoglobin - PubMedmaricela coqueNo ratings yet
- Module 03 Abo Blood Group SystemDocument8 pagesModule 03 Abo Blood Group SystemZoe DableNo ratings yet
- Hb Lepore disorder in a Malay familyDocument5 pagesHb Lepore disorder in a Malay familyimran ahmed siddiquiNo ratings yet
- Thalassaemia: Alpha Thalassaemia Beta Thalassaemia Delta-Beta ThalassaemiaDocument32 pagesThalassaemia: Alpha Thalassaemia Beta Thalassaemia Delta-Beta ThalassaemiaGovindaraju SubramaniNo ratings yet
- NotesDocument24 pagesNotesSapna JainNo ratings yet
- 7 Hemolytic Anemias PDFDocument44 pages7 Hemolytic Anemias PDFWin Ni del PîlarNo ratings yet
- Detection and Quantitation of Normal and Variant Haemoglobins: An Analytical ReviewDocument15 pagesDetection and Quantitation of Normal and Variant Haemoglobins: An Analytical ReviewAmanda MatthewsNo ratings yet
- Pallatto Et Al-2018-Veterinary Clinical PathologyDocument4 pagesPallatto Et Al-2018-Veterinary Clinical Pathologygranulomatous pneumoniaNo ratings yet
- 17395137artif CellsDocument18 pages17395137artif CellsIstván PortörőNo ratings yet
- ABCDocument56 pagesABCMary Kaye Yvonne OtillaNo ratings yet
- Thalassemia Syndromes: Hematology-Oncology Division Pediatric Departement School of Medicine University of North SumateraDocument34 pagesThalassemia Syndromes: Hematology-Oncology Division Pediatric Departement School of Medicine University of North SumateraAbdusSomadNo ratings yet
- Misleading Presentation of Haemoglobin ElectrophorDocument4 pagesMisleading Presentation of Haemoglobin Electrophornb8j9y7t8gNo ratings yet
- Gene Mutations & Sickle Celled AnaemiaDocument1 pageGene Mutations & Sickle Celled AnaemiaJohn OsborneNo ratings yet
- Toward 21st Century Blood Component Replacement Therapeutics: Artificial Oxygen Carriers, Platelet Substitutes, Recombinant Clotting Factors, and OthersDocument15 pagesToward 21st Century Blood Component Replacement Therapeutics: Artificial Oxygen Carriers, Platelet Substitutes, Recombinant Clotting Factors, and OthersIstván PortörőNo ratings yet
- Sickle Cell Diseases and Thalasemia Asma2Document60 pagesSickle Cell Diseases and Thalasemia Asma2Female calmNo ratings yet
- PB 0783Document9 pagesPB 0783Mona HelouNo ratings yet
- PKD BiochemistryDocument7 pagesPKD BiochemistryHyun Jae WonNo ratings yet
- 6 - HemoglobinopathiesDocument55 pages6 - HemoglobinopathiesSara BakerNo ratings yet
- Week 3: Hemoglobinopathies: HB Lepore Hbe Hbs HBC HB SC Disease HPFHDocument24 pagesWeek 3: Hemoglobinopathies: HB Lepore Hbe Hbs HBC HB SC Disease HPFHUdyani AgustinaNo ratings yet
- ABO Blood Group SystemDocument61 pagesABO Blood Group Systemmail2jackal50% (2)
- Hemoglobin-Vesicles As A Transfusion Alternative: Eishun Tsuchida and Hiromi SakaiDocument9 pagesHemoglobin-Vesicles As A Transfusion Alternative: Eishun Tsuchida and Hiromi SakaiIstván PortörőNo ratings yet
- Alpha-Thalassemia: Authors: Renzo Galanello, MD Antonio Cao, MDDocument31 pagesAlpha-Thalassemia: Authors: Renzo Galanello, MD Antonio Cao, MDKiran KumarNo ratings yet
- Quantitative Analysis of F Cells in Children With Sickle Cell DiseaseDocument7 pagesQuantitative Analysis of F Cells in Children With Sickle Cell DiseaseAraceli Enríquez OvandoNo ratings yet
- Development of Recombinant Hemoglobin-Based Oxygen Carriers: Orum Eview RticleDocument15 pagesDevelopment of Recombinant Hemoglobin-Based Oxygen Carriers: Orum Eview RticleJakeusNo ratings yet
- ABO Blood Group Discovery and GeneticsDocument64 pagesABO Blood Group Discovery and GeneticsAbhugz Marcelo100% (1)
- Thalassaemia S 1Document24 pagesThalassaemia S 1Nickesha MilletteNo ratings yet
- Interactions of hemoglobin Lepore (δβ hybrid hemoglobin) with various hemoglobinopathiesDocument6 pagesInteractions of hemoglobin Lepore (δβ hybrid hemoglobin) with various hemoglobinopathiesAndreia RibeiroNo ratings yet
- Thalassemias: Alissa Martin,, Alexis A. ThompsonDocument9 pagesThalassemias: Alissa Martin,, Alexis A. ThompsonAmada RosalesNo ratings yet
- Pediatric Thalassemia GuideDocument21 pagesPediatric Thalassemia GuideFenty IswarNo ratings yet
- Antibioticoterapia Local Versus Antibioticoterapia Sistémica For Desarrollo de Infección... 06-Mar-2021Document1 pageAntibioticoterapia Local Versus Antibioticoterapia Sistémica For Desarrollo de Infección... 06-Mar-2021Mauri ParadedaNo ratings yet
- Antibioticoterapia Local Versus Antibioticoterapia Sistémica For Desarrollo de Infección... 06-Mar-2021Document2 pagesAntibioticoterapia Local Versus Antibioticoterapia Sistémica For Desarrollo de Infección... 06-Mar-2021Mauri ParadedaNo ratings yet
- Damage Control Management y Polytrauma PatientDocument324 pagesDamage Control Management y Polytrauma PatientMauri ParadedaNo ratings yet
- Japanese Gastric Cancer Treatment 2014Document19 pagesJapanese Gastric Cancer Treatment 201409172216507No ratings yet
- Antibioticoterapia Local Versus Antibioticoterapia Sistémica For Desarrollo de Infección... 06-Mar-2021Document1 pageAntibioticoterapia Local Versus Antibioticoterapia Sistémica For Desarrollo de Infección... 06-Mar-2021Mauri ParadedaNo ratings yet
- Stroke in Young AdultsDocument8 pagesStroke in Young AdultsMauri ParadedaNo ratings yet
- Tumors and Tumor Like Lesions of BoneDocument963 pagesTumors and Tumor Like Lesions of BoneMauri Paradeda100% (1)
- Stroke Young CauseDocument9 pagesStroke Young CausefahlevyNo ratings yet
- chp:10.1007/978 94 011 4110 9 - 23Document8 pageschp:10.1007/978 94 011 4110 9 - 23Mauri ParadedaNo ratings yet
- Drug-Resistant Malaria - An InsightDocument14 pagesDrug-Resistant Malaria - An InsightMauri ParadedaNo ratings yet
- Malarial Hemozoin: From Target To ToolDocument27 pagesMalarial Hemozoin: From Target To ToolMauri ParadedaNo ratings yet
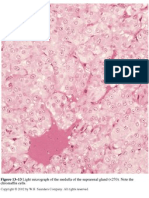
- Figure 13-13 Light Micrograph of The Medulla of The Suprarenal Gland (Document1 pageFigure 13-13 Light Micrograph of The Medulla of The Suprarenal Gland (Mauri ParadedaNo ratings yet
- The Human Oral Microbiome † 储Document16 pagesThe Human Oral Microbiome † 储Sebastian NegrutNo ratings yet
- Industrial Biotechnology 2Document12 pagesIndustrial Biotechnology 2Techno India UniversityNo ratings yet
- Gene IsolationDocument25 pagesGene Isolationrag.1607No ratings yet
- Corning Micro Array TechnologyDocument19 pagesCorning Micro Array Technologytalk2arfat50% (2)
- Introduction To Cell Biology 2010-2011 FinalDocument16 pagesIntroduction To Cell Biology 2010-2011 FinalMatthew GuoNo ratings yet
- Lab Investigation AP 3 - Constructing Evol Lineages With DNA BLASTDocument7 pagesLab Investigation AP 3 - Constructing Evol Lineages With DNA BLASTKeri Gobin SamarooNo ratings yet
- Medical Genomics and Metagenomics Hands-on TrainingDocument1 pageMedical Genomics and Metagenomics Hands-on TrainingManjunath MNo ratings yet
- Virus WorksheetDocument8 pagesVirus WorksheetAnisa S. AlfianaNo ratings yet
- Technological Advancement EssayDocument3 pagesTechnological Advancement EssayHania AliNo ratings yet
- MSC Biotechnology Second Year AssignmentsDocument2 pagesMSC Biotechnology Second Year AssignmentsAISHA MUHAMMADNo ratings yet
- Biochemistry Gat CourseDocument11 pagesBiochemistry Gat Courseki_dvm100% (1)
- Top 10 Leading Bio XXXDocument8 pagesTop 10 Leading Bio XXXSyed ShoyebNo ratings yet
- Chemical Engineering . A Career For You: 8/20/2011 UH Chem. Eng. Dept. 1Document24 pagesChemical Engineering . A Career For You: 8/20/2011 UH Chem. Eng. Dept. 1Trunk DangNo ratings yet
- GPB SyllabusDocument27 pagesGPB SyllabusYASHPAL SINGH100% (1)
- Chapter 13.4 & 15.4 Active Reading GuideDocument5 pagesChapter 13.4 & 15.4 Active Reading GuidedorothyNo ratings yet
- Fundamentals of DNA-Chip - Array TechnologyDocument5 pagesFundamentals of DNA-Chip - Array TechnologyvarinderkumarscribdNo ratings yet
- Career Guidance For 10th STDDocument50 pagesCareer Guidance For 10th STDvijayalakshmiramanNo ratings yet
- DisposableDocument9 pagesDisposableSuzanne TaylorNo ratings yet
- Induced Pluripotent Stem Cells in Medicine and BiologyDocument5 pagesInduced Pluripotent Stem Cells in Medicine and BiologyMaria Del Mar Robles100% (1)
- Top 100 Pharma CompaniesDocument3 pagesTop 100 Pharma CompaniesApurva Ranjan100% (1)
- Building Biotechnology 3rd EditionDocument24 pagesBuilding Biotechnology 3rd EditionPrincess Anne Lantin0% (1)
- Transposition: A DNA Recombination Reaction that Results in TranslocationDocument42 pagesTransposition: A DNA Recombination Reaction that Results in TranslocationToh Qin KaneNo ratings yet
- Bio-Medical Waste Rules GuideDocument19 pagesBio-Medical Waste Rules GuideAruna ChezhianNo ratings yet
- Cell Theory Scientists Types Reinforcement WorksheetDocument2 pagesCell Theory Scientists Types Reinforcement Worksheetapi-165869000No ratings yet
- Sachin Kumar CVDocument3 pagesSachin Kumar CVSachin KumarNo ratings yet
- Abbvie Inc. earnings and valuationDocument1 pageAbbvie Inc. earnings and valuationderek_2010No ratings yet
- Presentation Foamed Concrete 2009Document25 pagesPresentation Foamed Concrete 2009Mukut DasNo ratings yet
- Unit 6 - BioinformaticsDocument41 pagesUnit 6 - BioinformaticsLeonNo ratings yet
- Genetika Dan Ekspresi Gen Pada EukariotDocument36 pagesGenetika Dan Ekspresi Gen Pada EukariotRaditya Indah TofaniNo ratings yet
- SAFC Biosciences - Technical Bulletin - EX-CELL™ Vero Serum-Free Medium For High-Density Vero GrowthDocument4 pagesSAFC Biosciences - Technical Bulletin - EX-CELL™ Vero Serum-Free Medium For High-Density Vero GrowthSAFC-GlobalNo ratings yet