You might also like
- Toxicological Effects of Pyret On Non-Target Aquatic Insects 3Document1 pageToxicological Effects of Pyret On Non-Target Aquatic Insects 3Angel NorabuenaNo ratings yet
- Toxicological Effects of Pyret On Non-Target Aquatic Insects 7Document1 pageToxicological Effects of Pyret On Non-Target Aquatic Insects 7Angel NorabuenaNo ratings yet
- Toxicological Effects of Pyret On Non-Target Aquatic Insects 5Document1 pageToxicological Effects of Pyret On Non-Target Aquatic Insects 5Angel NorabuenaNo ratings yet
- Geoderma: Mohammad Sadegh Askari, Sharon M. O'Rourke, Nicholas M. HoldenDocument12 pagesGeoderma: Mohammad Sadegh Askari, Sharon M. O'Rourke, Nicholas M. HoldenAngel NorabuenaNo ratings yet
- Coa Mix 1 PDFDocument4 pagesCoa Mix 1 PDFAngel NorabuenaNo ratings yet
- Arsenico en AguasDocument3 pagesArsenico en AguasAngel Norabuena100% (1)
- Shoe Dog: A Memoir by the Creator of NikeFrom EverandShoe Dog: A Memoir by the Creator of NikeRating: 4.5 out of 5 stars4.5/5 (537)
- The Yellow House: A Memoir (2019 National Book Award Winner)From EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Rating: 4 out of 5 stars4/5 (98)
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeFrom EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeRating: 4 out of 5 stars4/5 (5794)
- The Little Book of Hygge: Danish Secrets to Happy LivingFrom EverandThe Little Book of Hygge: Danish Secrets to Happy LivingRating: 3.5 out of 5 stars3.5/5 (400)
- Grit: The Power of Passion and PerseveranceFrom EverandGrit: The Power of Passion and PerseveranceRating: 4 out of 5 stars4/5 (588)
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureFrom EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureRating: 4.5 out of 5 stars4.5/5 (474)
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryFrom EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryRating: 3.5 out of 5 stars3.5/5 (231)
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceFrom EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceRating: 4 out of 5 stars4/5 (895)
- Team of Rivals: The Political Genius of Abraham LincolnFrom EverandTeam of Rivals: The Political Genius of Abraham LincolnRating: 4.5 out of 5 stars4.5/5 (234)
- Never Split the Difference: Negotiating As If Your Life Depended On ItFrom EverandNever Split the Difference: Negotiating As If Your Life Depended On ItRating: 4.5 out of 5 stars4.5/5 (838)
- The Emperor of All Maladies: A Biography of CancerFrom EverandThe Emperor of All Maladies: A Biography of CancerRating: 4.5 out of 5 stars4.5/5 (271)
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaFrom EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaRating: 4.5 out of 5 stars4.5/5 (266)
- On Fire: The (Burning) Case for a Green New DealFrom EverandOn Fire: The (Burning) Case for a Green New DealRating: 4 out of 5 stars4/5 (74)
- The Unwinding: An Inner History of the New AmericaFrom EverandThe Unwinding: An Inner History of the New AmericaRating: 4 out of 5 stars4/5 (45)
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersFrom EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersRating: 4.5 out of 5 stars4.5/5 (345)
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyFrom EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyRating: 3.5 out of 5 stars3.5/5 (2259)
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreFrom EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreRating: 4 out of 5 stars4/5 (1090)
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)From EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Rating: 4.5 out of 5 stars4.5/5 (121)
- Her Body and Other Parties: StoriesFrom EverandHer Body and Other Parties: StoriesRating: 4 out of 5 stars4/5 (821)
- An Efficient Algorithm For The Calculation of A Constant Q TransformDocument4 pagesAn Efficient Algorithm For The Calculation of A Constant Q TransformKarlAschnikowNo ratings yet
- PhysicsDocument396 pagesPhysicsGreeny34No ratings yet
- TSL250R, TSL251R, TSL252R: Light-to-Voltage Optical SensorsDocument21 pagesTSL250R, TSL251R, TSL252R: Light-to-Voltage Optical Sensorsthevincenzo@gmail.comNo ratings yet
- TIMSS8 Science ConceptsItems 6Document25 pagesTIMSS8 Science ConceptsItems 6adisan777No ratings yet
- Chapter 7 - Quantization Noise in DSPDocument31 pagesChapter 7 - Quantization Noise in DSPSarah KumakoNo ratings yet
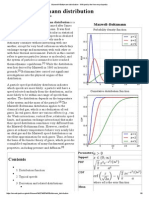
- Maxwell-Boltzmann DistributionDocument8 pagesMaxwell-Boltzmann DistributionlamyantingNo ratings yet
- Solar Tracker Using ArduinoDocument3 pagesSolar Tracker Using ArduinoInternational Journal of Innovative Science and Research Technology100% (1)
- Introduction To Linear MeasurementDocument6 pagesIntroduction To Linear MeasurementLalali LiNo ratings yet
- tp5 Mixed Convection With 2 Movable WallsDocument6 pagestp5 Mixed Convection With 2 Movable WallsTahar ADJOUDJNo ratings yet
- Buffer SelectionDocument3 pagesBuffer SelectionSandeep KumarNo ratings yet
- RRRRRRRRRRRRRRRRRRRRRRRRRRRRRRRRRRRRRRRRRRRRRRRRRRRRRRDocument14 pagesRRRRRRRRRRRRRRRRRRRRRRRRRRRRRRRRRRRRRRRRRRRRRRRRRRRRRRPranay GoswamiNo ratings yet
- Assignment Top Sheet Department of Civil Engineering & TechnologyDocument6 pagesAssignment Top Sheet Department of Civil Engineering & TechnologyEngr MahwishNo ratings yet
- Machining PDFDocument5 pagesMachining PDFlambrosNo ratings yet
- G. ACI 360R-06 Brings Slabs On Ground Into The 21st Century - Art McKinney PDFDocument2 pagesG. ACI 360R-06 Brings Slabs On Ground Into The 21st Century - Art McKinney PDFinitbashNo ratings yet
- 1.10.0-.Calculo de Muros de Contencion PDFDocument304 pages1.10.0-.Calculo de Muros de Contencion PDFJose Antonio Paredes VeraNo ratings yet
- Soil Test SidipurDocument22 pagesSoil Test SidipurRajib MaharjanNo ratings yet
- Materials and Testing MethodsDocument19 pagesMaterials and Testing MethodsWai Yann ZawNo ratings yet
- 16 TewariDocument22 pages16 TewariNebojsa BascarevicNo ratings yet
- Fatigue Failure Analysis of Fillet Welded Joints Used in Offshore StructuresDocument78 pagesFatigue Failure Analysis of Fillet Welded Joints Used in Offshore Structureschrism225836No ratings yet
- 10aug 2015 VJTIDocument34 pages10aug 2015 VJTIAyush Saxena100% (1)
- CHE122 Engineering Chemistry Laboratory 16842::Dr. Tanay Pramanik 0.0 0.0 2.0 1.0 1:discipline Knowledge, 2:skill EnhancementDocument5 pagesCHE122 Engineering Chemistry Laboratory 16842::Dr. Tanay Pramanik 0.0 0.0 2.0 1.0 1:discipline Knowledge, 2:skill EnhancementSandeep KakranNo ratings yet
- Oceanic Anoxic Events (O.a.e) Organic Rocks Deposition CretaceousDocument6 pagesOceanic Anoxic Events (O.a.e) Organic Rocks Deposition CretaceousJulian De Bedout OrdoñezNo ratings yet
- Abbe Refraktometer Carl Zeiss Jena - Type G 08 PDFDocument8 pagesAbbe Refraktometer Carl Zeiss Jena - Type G 08 PDFrowanrowanNo ratings yet
- Light and Shadow Quiz NameDocument4 pagesLight and Shadow Quiz Nameapi-490528795100% (1)
- Intro Electromechanisms Devices InstructorDocument76 pagesIntro Electromechanisms Devices Instructormatang_lawinNo ratings yet
- An Introduction To Numerical Methods For The Solutions of Partial Differential EquationsDocument12 pagesAn Introduction To Numerical Methods For The Solutions of Partial Differential EquationseiroNo ratings yet
- B Splines 04 PDFDocument16 pagesB Splines 04 PDFShawn PetersenNo ratings yet
- 8.6C PosttestDocument2 pages8.6C PosttestYohanes RatnodiyantoNo ratings yet
- Soln1 PsDocument6 pagesSoln1 PsRafran Rosly100% (1)
- PerformanceDocument14 pagesPerformancealmadhagiNo ratings yet