You might also like
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)From EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Rating: 4.5 out of 5 stars4.5/5 (119)
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaFrom EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaRating: 4.5 out of 5 stars4.5/5 (265)
- The Little Book of Hygge: Danish Secrets to Happy LivingFrom EverandThe Little Book of Hygge: Danish Secrets to Happy LivingRating: 3.5 out of 5 stars3.5/5 (399)
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryFrom EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryRating: 3.5 out of 5 stars3.5/5 (231)
- Grit: The Power of Passion and PerseveranceFrom EverandGrit: The Power of Passion and PerseveranceRating: 4 out of 5 stars4/5 (587)
- Never Split the Difference: Negotiating As If Your Life Depended On ItFrom EverandNever Split the Difference: Negotiating As If Your Life Depended On ItRating: 4.5 out of 5 stars4.5/5 (838)
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeFrom EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeRating: 4 out of 5 stars4/5 (5794)
- Team of Rivals: The Political Genius of Abraham LincolnFrom EverandTeam of Rivals: The Political Genius of Abraham LincolnRating: 4.5 out of 5 stars4.5/5 (234)
- Shoe Dog: A Memoir by the Creator of NikeFrom EverandShoe Dog: A Memoir by the Creator of NikeRating: 4.5 out of 5 stars4.5/5 (537)
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyFrom EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyRating: 3.5 out of 5 stars3.5/5 (2219)
- The Emperor of All Maladies: A Biography of CancerFrom EverandThe Emperor of All Maladies: A Biography of CancerRating: 4.5 out of 5 stars4.5/5 (271)
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreFrom EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreRating: 4 out of 5 stars4/5 (1090)
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersFrom EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersRating: 4.5 out of 5 stars4.5/5 (344)
- Her Body and Other Parties: StoriesFrom EverandHer Body and Other Parties: StoriesRating: 4 out of 5 stars4/5 (821)
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceFrom EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceRating: 4 out of 5 stars4/5 (894)
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureFrom EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureRating: 4.5 out of 5 stars4.5/5 (474)
- The Unwinding: An Inner History of the New AmericaFrom EverandThe Unwinding: An Inner History of the New AmericaRating: 4 out of 5 stars4/5 (45)
- The Yellow House: A Memoir (2019 National Book Award Winner)From EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Rating: 4 out of 5 stars4/5 (98)
- On Fire: The (Burning) Case for a Green New DealFrom EverandOn Fire: The (Burning) Case for a Green New DealRating: 4 out of 5 stars4/5 (73)
- Re CessnaSingle 1996on Structural Repair MM SESR04Document167 pagesRe CessnaSingle 1996on Structural Repair MM SESR04chipocludo7av2100% (4)
- Experimental Lab Report 1Document10 pagesExperimental Lab Report 1api-274857931100% (10)
- Boylestad FET AmplDocument64 pagesBoylestad FET AmplUltrazordNo ratings yet
- Welding Defects and PreventionDocument2 pagesWelding Defects and PreventionVicky SinghNo ratings yet
- BK TNG 001 TS PI SP 001 - Piping Specification - Rev 2Document45 pagesBK TNG 001 TS PI SP 001 - Piping Specification - Rev 2Kev TraNo ratings yet
- 40 - The Quantized Electromagnetic Field PDFDocument21 pages40 - The Quantized Electromagnetic Field PDFUltrazordNo ratings yet
- Controlling Wax Deposition Presence Hydrates-01!25!10Document85 pagesControlling Wax Deposition Presence Hydrates-01!25!10wjawichNo ratings yet
- Biotechnology Reviewer - Photosynthesis and Cellular RespirationDocument3 pagesBiotechnology Reviewer - Photosynthesis and Cellular RespirationMaribeth VillanuevaNo ratings yet
- Subject Index Final 30nov16Document33 pagesSubject Index Final 30nov16UltrazordNo ratings yet
- Parts Index Final 30nov16Document23 pagesParts Index Final 30nov16UltrazordNo ratings yet
- 45 - Lorentz Transformations in Special Relativity (Incomplete)Document19 pages45 - Lorentz Transformations in Special Relativity (Incomplete)UltrazordNo ratings yet
- 42 - Scattering of Radiation (Incomplete) PDFDocument7 pages42 - Scattering of Radiation (Incomplete) PDFUltrazordNo ratings yet
- Appendix E - Tensor AnalysisDocument19 pagesAppendix E - Tensor AnalysisUltrazordNo ratings yet
- 41 - Interaction of Radiation With Matter PDFDocument19 pages41 - Interaction of Radiation With Matter PDFUltrazordNo ratings yet
- Appendix C - Gaussian IntegralsDocument3 pagesAppendix C - Gaussian IntegralsUltrazordNo ratings yet
- Appendix D - Vector CalculusDocument3 pagesAppendix D - Vector CalculusUltrazordNo ratings yet
- 46 - Covariance of The Dirac Equation (Incomplete)Document6 pages46 - Covariance of The Dirac Equation (Incomplete)UltrazordNo ratings yet
- 43 - The Klein-Gordon Equation PDFDocument7 pages43 - The Klein-Gordon Equation PDFUltrazordNo ratings yet
- 44 - Introduction To The Dirac Equation (Incomplete)Document13 pages44 - Introduction To The Dirac Equation (Incomplete)UltrazordNo ratings yet
- 39 - Lagrangian and Hamiltonian Formulation of The Classical Electromagnetic Field PDFDocument27 pages39 - Lagrangian and Hamiltonian Formulation of The Classical Electromagnetic Field PDFUltrazordNo ratings yet
- 36 - The Lippmann-Schwinger Equation PDFDocument16 pages36 - The Lippmann-Schwinger Equation PDFUltrazordNo ratings yet
- 37 - Adiabatic Invariance, The Geometric Phase, and The Born-Oppenheimer Approximation PDFDocument12 pages37 - Adiabatic Invariance, The Geometric Phase, and The Born-Oppenheimer Approximation PDFUltrazordNo ratings yet
- 35 - Green's Functions in Quantum Mechanics PDFDocument23 pages35 - Green's Functions in Quantum Mechanics PDFUltrazordNo ratings yet
- 38 - The Classical Electromagnetic Field Hamiltonian PDFDocument29 pages38 - The Classical Electromagnetic Field Hamiltonian PDFUltrazordNo ratings yet
- Introduction to Scattering Theory and Potential ScatteringDocument16 pagesIntroduction to Scattering Theory and Potential ScatteringUltrazordNo ratings yet
- 33 - The Photoelectric Effect PDFDocument9 pages33 - The Photoelectric Effect PDFUltrazordNo ratings yet
- 29 - The Thomas-Fermi Model PDFDocument9 pages29 - The Thomas-Fermi Model PDFUltrazordNo ratings yet
- 32 - Time-Dependent Perturbation Theory PDFDocument25 pages32 - Time-Dependent Perturbation Theory PDFUltrazordNo ratings yet
- 26 - The Variational Method PDFDocument7 pages26 - The Variational Method PDFUltrazordNo ratings yet
- 31 - Elements of Atomic Structure in Multi-Electron Atoms PDFDocument6 pages31 - Elements of Atomic Structure in Multi-Electron Atoms PDFUltrazordNo ratings yet
- 28 - Helium and Helium-Like Atoms PDFDocument26 pages28 - Helium and Helium-Like Atoms PDFUltrazordNo ratings yet
- Fine structure in hydrogen and alkali atomsDocument15 pagesFine structure in hydrogen and alkali atomsUltrazordNo ratings yet
- 30 - The Hartree-Fock Method in Atoms PDFDocument29 pages30 - The Hartree-Fock Method in Atoms PDFUltrazordNo ratings yet
- Physics 221A Notes on Hyperfine StructureDocument12 pagesPhysics 221A Notes on Hyperfine StructureUltrazord100% (1)
- 24 - The Zeeman Effect in Hydrogen and Alkali Atoms PDFDocument11 pages24 - The Zeeman Effect in Hydrogen and Alkali Atoms PDFUltrazordNo ratings yet
- B.pharm. Class NotesDocument817 pagesB.pharm. Class NotesMukesh TiwariNo ratings yet
- CHEM 18 PROBLEM SET CHEMICAL THERMODYNAMICS AND EQUILIBRIUMDocument4 pagesCHEM 18 PROBLEM SET CHEMICAL THERMODYNAMICS AND EQUILIBRIUMDaniel Jann CotiaNo ratings yet
- Chemistry, Mathematics & Physics: All India Internal Test SeriesDocument15 pagesChemistry, Mathematics & Physics: All India Internal Test Seriesmadhav aggarwalNo ratings yet
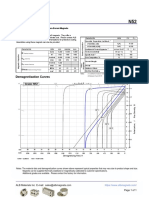
- N52 Grade Neodymium Magnets DataDocument1 pageN52 Grade Neodymium Magnets DataSteve HsuNo ratings yet
- AWS D1.5 PQR TitleDocument1 pageAWS D1.5 PQR TitleNavanitheeshwaran SivasubramaniyamNo ratings yet
- Msds Icr 122 LSFDocument12 pagesMsds Icr 122 LSFWisnu AbaraiNo ratings yet
- Vortex Quantum SeriesDocument34 pagesVortex Quantum SeriesmiguelcNo ratings yet
- Ammonia Synthesis Process OverviewDocument31 pagesAmmonia Synthesis Process OverviewKhalid AkNo ratings yet
- CHE211 Problem Set 5Document3 pagesCHE211 Problem Set 5AlexNo ratings yet
- Heavy Oil Upgrading - A Key Solution For Heavy Oil Upstream and Midstream Operations - IVANHOE ENERGY PDFDocument44 pagesHeavy Oil Upgrading - A Key Solution For Heavy Oil Upstream and Midstream Operations - IVANHOE ENERGY PDFGustavo Gonzalez ServaNo ratings yet
- Global Desiccants and Adsorbents MarketDocument5 pagesGlobal Desiccants and Adsorbents MarketPulkit BatraNo ratings yet
- 11 Numerical AnalysisDocument9 pages11 Numerical Analysisعزالدين حسنNo ratings yet
- Factors Affecting Rate of EvaporationDocument22 pagesFactors Affecting Rate of EvaporationShimnu MoneNo ratings yet
- Ankit Topic - Using Cast Iron For Machine PartsDocument12 pagesAnkit Topic - Using Cast Iron For Machine PartsAnkit BhadesiaNo ratings yet
- Math 6 Unit 8 Volume of Solids and LiquidsDocument1 pageMath 6 Unit 8 Volume of Solids and LiquidsRobi AkmalNo ratings yet
- BDA30603 Tutorial 4Document7 pagesBDA30603 Tutorial 4Firdaus JannahNo ratings yet
- Compressor Disk Corrosion Problems and Solutions - SermetelDocument13 pagesCompressor Disk Corrosion Problems and Solutions - SermetelKatNo ratings yet
- Laboratory Exercise 4Document3 pagesLaboratory Exercise 4dennise reyesNo ratings yet
- Mark Scheme (Results) Summer 2015: GCE Chemistry (6CH01/01) The Core Principles of ChemistryDocument21 pagesMark Scheme (Results) Summer 2015: GCE Chemistry (6CH01/01) The Core Principles of ChemistryAmeenIbrahimNo ratings yet
- B705 Acpt1003Document3 pagesB705 Acpt1003taya1401No ratings yet
- What's New - PV Elite 2018Document28 pagesWhat's New - PV Elite 2018SathiyaseelanNo ratings yet
- Us03cicv21 Unit3Document28 pagesUs03cicv21 Unit3ashokNo ratings yet
- Recycling of Pad-Batch Washing Textile Wastewater Through Advanced Oxidation Processes and Its Reusability Assessment For Turkish Textile IndustDocument7 pagesRecycling of Pad-Batch Washing Textile Wastewater Through Advanced Oxidation Processes and Its Reusability Assessment For Turkish Textile IndustGizem D.No ratings yet
- Ion Exchange PDFDocument18 pagesIon Exchange PDFSarah LimaNo ratings yet