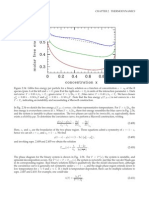
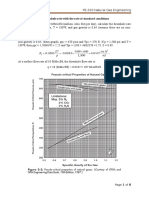
You might also like
- Obama BudgetDocument5 pagesObama BudgetVictor EnemNo ratings yet
- 2015 Drinking 1Document1 page2015 Drinking 1Victor EnemNo ratings yet
- Alcohol Facts and Statistics: Alcohol Use in The United StatesDocument6 pagesAlcohol Facts and Statistics: Alcohol Use in The United StatesVictor EnemNo ratings yet
- N 400Document20 pagesN 400Leo FerrNo ratings yet
- Suicide Datasheet-Facts at A Glance-2015Document2 pagesSuicide Datasheet-Facts at A Glance-2015takemebrickNo ratings yet
- Fy2013 Budget RolloutDocument25 pagesFy2013 Budget RolloutVictor EnemNo ratings yet
- An Introduction To Fluid Mechanics: Notes For The First Year Lecture CourseDocument5 pagesAn Introduction To Fluid Mechanics: Notes For The First Year Lecture CourseVictor EnemNo ratings yet
- Financial AccountingDocument3 pagesFinancial AccountingVictor EnemNo ratings yet
- Fy2013 Fast Facts VAs Budget HighlightsDocument1 pageFy2013 Fast Facts VAs Budget HighlightsVictor EnemNo ratings yet
- Chapter 2. Thermodynamics: S, N P P, N V P T, NDocument10 pagesChapter 2. Thermodynamics: S, N P P, N V P T, NVictor EnemNo ratings yet
- Fy2013 Budget RolloutDocument25 pagesFy2013 Budget RolloutVictor EnemNo ratings yet
- Whitepap ThermoDocument8 pagesWhitepap ThermoVictor EnemNo ratings yet
- Solution Manual NotesDocument5 pagesSolution Manual NotesVictor Enem9% (11)
- Thermo Lecture NotesDocument10 pagesThermo Lecture NotesVictor EnemNo ratings yet
- Thermo Chapter ElevenDocument10 pagesThermo Chapter ElevenVictor EnemNo ratings yet
- Thermo Chapter TvelveDocument10 pagesThermo Chapter TvelveVictor EnemNo ratings yet
- Thermo Chapter NineDocument10 pagesThermo Chapter NineVictor EnemNo ratings yet
- Thermo Chapter OneDocument16 pagesThermo Chapter OneVictor EnemNo ratings yet
- Chapter 2. Thermodynamics: 2.11.5 Joule Effect: Free Expansion of A GasDocument10 pagesChapter 2. Thermodynamics: 2.11.5 Joule Effect: Free Expansion of A GasVictor EnemNo ratings yet
- Thermo Chapter FiveDocument10 pagesThermo Chapter FiveVictor EnemNo ratings yet
- Thermo Chapter FourDocument10 pagesThermo Chapter FourVictor EnemNo ratings yet
- Thermo Chapter SixDocument10 pagesThermo Chapter SixVictor EnemNo ratings yet
- Thermodynamic Variables Versus Microscopic VariablesDocument6 pagesThermodynamic Variables Versus Microscopic Variablesanon_251774No ratings yet
- Thermo Chapter ThreeDocument10 pagesThermo Chapter ThreeVictor EnemNo ratings yet
- Thermo Chapter TwoDocument9 pagesThermo Chapter TwoVictor EnemNo ratings yet
- 2015 Holiday Processing Schedule For Receivables Operations (Lockbox)Document2 pages2015 Holiday Processing Schedule For Receivables Operations (Lockbox)Victor EnemNo ratings yet
- Thermodynamics Refrigerator NotesDocument35 pagesThermodynamics Refrigerator NotesVictor EnemNo ratings yet
- Never Split the Difference: Negotiating As If Your Life Depended On ItFrom EverandNever Split the Difference: Negotiating As If Your Life Depended On ItRating: 4.5 out of 5 stars4.5/5 (838)
- Shoe Dog: A Memoir by the Creator of NikeFrom EverandShoe Dog: A Memoir by the Creator of NikeRating: 4.5 out of 5 stars4.5/5 (537)
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeFrom EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeRating: 4 out of 5 stars4/5 (5794)
- The Yellow House: A Memoir (2019 National Book Award Winner)From EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Rating: 4 out of 5 stars4/5 (98)
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceFrom EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceRating: 4 out of 5 stars4/5 (894)
- The Little Book of Hygge: Danish Secrets to Happy LivingFrom EverandThe Little Book of Hygge: Danish Secrets to Happy LivingRating: 3.5 out of 5 stars3.5/5 (399)
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureFrom EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureRating: 4.5 out of 5 stars4.5/5 (474)
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryFrom EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryRating: 3.5 out of 5 stars3.5/5 (231)
- Grit: The Power of Passion and PerseveranceFrom EverandGrit: The Power of Passion and PerseveranceRating: 4 out of 5 stars4/5 (587)
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaFrom EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaRating: 4.5 out of 5 stars4.5/5 (265)
- On Fire: The (Burning) Case for a Green New DealFrom EverandOn Fire: The (Burning) Case for a Green New DealRating: 4 out of 5 stars4/5 (73)
- The Emperor of All Maladies: A Biography of CancerFrom EverandThe Emperor of All Maladies: A Biography of CancerRating: 4.5 out of 5 stars4.5/5 (271)
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersFrom EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersRating: 4.5 out of 5 stars4.5/5 (344)
- The Unwinding: An Inner History of the New AmericaFrom EverandThe Unwinding: An Inner History of the New AmericaRating: 4 out of 5 stars4/5 (45)
- Team of Rivals: The Political Genius of Abraham LincolnFrom EverandTeam of Rivals: The Political Genius of Abraham LincolnRating: 4.5 out of 5 stars4.5/5 (234)
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyFrom EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyRating: 3.5 out of 5 stars3.5/5 (2219)
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreFrom EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreRating: 4 out of 5 stars4/5 (1090)
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)From EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Rating: 4.5 out of 5 stars4.5/5 (119)
- Her Body and Other Parties: StoriesFrom EverandHer Body and Other Parties: StoriesRating: 4 out of 5 stars4/5 (821)
- 8 Lightning CalculationDocument75 pages8 Lightning CalculationSamant SauravNo ratings yet
- Example 3 Relating Downhole Rate With The Rate at Standard ConditionsDocument8 pagesExample 3 Relating Downhole Rate With The Rate at Standard ConditionsMaisam AbbasNo ratings yet
- MCE433 Advanced Thermodynamics (HOD-DeAN)Document11 pagesMCE433 Advanced Thermodynamics (HOD-DeAN)KEHINDE BABALOLANo ratings yet
- Ongc Hazira Plant A Techno Commercial Insight'Document21 pagesOngc Hazira Plant A Techno Commercial Insight'Bhavesh PatelNo ratings yet
- SPD Characteristics For Equipment Protection: ICLP 2010Document8 pagesSPD Characteristics For Equipment Protection: ICLP 2010Zainal KadirNo ratings yet
- Module 3 TorsionDocument8 pagesModule 3 TorsionGeri LazaroNo ratings yet
- What Is A Buck Boost ConverterDocument4 pagesWhat Is A Buck Boost ConverterXavierNo ratings yet
- GBT 7894-2001fundamental Technical Requirements For Hydro-GeneratorsDocument20 pagesGBT 7894-2001fundamental Technical Requirements For Hydro-Generatorsaji.isramboNo ratings yet
- Refrigeration Cycle ExperimentDocument10 pagesRefrigeration Cycle ExperimentJay NgNo ratings yet
- Summer Gizmo Lab 2Document4 pagesSummer Gizmo Lab 2Liam HoustonNo ratings yet
- BLDC COMPRESSOR TCC DA HORIZONTAL r1.0Document18 pagesBLDC COMPRESSOR TCC DA HORIZONTAL r1.0Bruno Souza100% (2)
- Broadband Nonreciprocal Thermal Radiation With Weyl Semimetal-Based Pattern-Free HeterostructureDocument4 pagesBroadband Nonreciprocal Thermal Radiation With Weyl Semimetal-Based Pattern-Free HeterostructureGergő BárdosNo ratings yet
- Resistors in Series and Parallel PDFDocument7 pagesResistors in Series and Parallel PDFEJ CelestialNo ratings yet
- NAPTIN Graduate Skills Programme BrochureDocument40 pagesNAPTIN Graduate Skills Programme BrochureEngr KushenscottNo ratings yet
- Universiti Malaysia Pahang: Faculty of Mechanical EngineeringDocument13 pagesUniversiti Malaysia Pahang: Faculty of Mechanical EngineeringFirdaus IliasNo ratings yet
- MCQ Res1Document6 pagesMCQ Res1Durairaj M.No ratings yet
- Question Paper of Engineering Service Examination 2010 Electrical Engineering Paper-IDocument22 pagesQuestion Paper of Engineering Service Examination 2010 Electrical Engineering Paper-ISantosh SandyNo ratings yet
- Test Certificate N° 1452847 Project 908293-01: Mesured Voltage RatioDocument1 pageTest Certificate N° 1452847 Project 908293-01: Mesured Voltage RatioMardiansyah AtrimaNo ratings yet
- Problem 10.7Document3 pagesProblem 10.7台師大陳彥穎No ratings yet
- NEPLAN Turbine-Governor ModelsDocument99 pagesNEPLAN Turbine-Governor ModelsRatilal M Jadav100% (1)
- MCC Bus Bar Sizing CalculationDocument42 pagesMCC Bus Bar Sizing Calculationmohan babuNo ratings yet
- Question bank-FYBScDocument8 pagesQuestion bank-FYBScUnknown AmanNo ratings yet
- Virtual Work Analysis of Mechanical Systems in EquilibriumDocument20 pagesVirtual Work Analysis of Mechanical Systems in EquilibriumarslansaeedarslanNo ratings yet
- GENPHY2 - Lesson - Ohm's Law and ResistanceDocument3 pagesGENPHY2 - Lesson - Ohm's Law and ResistanceJazmaine SimbulanNo ratings yet
- High Voltage Power Supply: Technical LeafletDocument12 pagesHigh Voltage Power Supply: Technical LeafletDavid_Allen_007No ratings yet
- 300/500ma Low Dropout Linear Voltage Regulator: General Description FeaturesDocument11 pages300/500ma Low Dropout Linear Voltage Regulator: General Description FeaturesPedro RodriguezNo ratings yet
- Mil HDBK 978B PDFDocument1,224 pagesMil HDBK 978B PDFjsadachiNo ratings yet
- Em 123Document76 pagesEm 123Naresh DamaNo ratings yet
- June 2014 (R) QP - Unit 1 Edexcel Physics A-LevelDocument28 pagesJune 2014 (R) QP - Unit 1 Edexcel Physics A-LevelArun MosesNo ratings yet
- IkhideMA - PHD Thesis PDFDocument247 pagesIkhideMA - PHD Thesis PDFfsdgsgsNo ratings yet