You might also like
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryFrom EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryRating: 3.5 out of 5 stars3.5/5 (231)
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)From EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Rating: 4.5 out of 5 stars4.5/5 (121)
- Grit: The Power of Passion and PerseveranceFrom EverandGrit: The Power of Passion and PerseveranceRating: 4 out of 5 stars4/5 (588)
- Never Split the Difference: Negotiating As If Your Life Depended On ItFrom EverandNever Split the Difference: Negotiating As If Your Life Depended On ItRating: 4.5 out of 5 stars4.5/5 (838)
- The Little Book of Hygge: Danish Secrets to Happy LivingFrom EverandThe Little Book of Hygge: Danish Secrets to Happy LivingRating: 3.5 out of 5 stars3.5/5 (400)
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaFrom EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaRating: 4.5 out of 5 stars4.5/5 (266)
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeFrom EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeRating: 4 out of 5 stars4/5 (5794)
- Her Body and Other Parties: StoriesFrom EverandHer Body and Other Parties: StoriesRating: 4 out of 5 stars4/5 (821)
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreFrom EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreRating: 4 out of 5 stars4/5 (1090)
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyFrom EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyRating: 3.5 out of 5 stars3.5/5 (2259)
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersFrom EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersRating: 4.5 out of 5 stars4.5/5 (345)
- Shoe Dog: A Memoir by the Creator of NikeFrom EverandShoe Dog: A Memoir by the Creator of NikeRating: 4.5 out of 5 stars4.5/5 (537)
- The Emperor of All Maladies: A Biography of CancerFrom EverandThe Emperor of All Maladies: A Biography of CancerRating: 4.5 out of 5 stars4.5/5 (271)
- Team of Rivals: The Political Genius of Abraham LincolnFrom EverandTeam of Rivals: The Political Genius of Abraham LincolnRating: 4.5 out of 5 stars4.5/5 (234)
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceFrom EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceRating: 4 out of 5 stars4/5 (895)
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureFrom EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureRating: 4.5 out of 5 stars4.5/5 (474)
- On Fire: The (Burning) Case for a Green New DealFrom EverandOn Fire: The (Burning) Case for a Green New DealRating: 4 out of 5 stars4/5 (74)
- 06f Engine CFM LEAP NEO Control and Indication Rev 02Document35 pages06f Engine CFM LEAP NEO Control and Indication Rev 02Ricardo Rubio92% (12)
- The Yellow House: A Memoir (2019 National Book Award Winner)From EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Rating: 4 out of 5 stars4/5 (98)
- The Unwinding: An Inner History of the New AmericaFrom EverandThe Unwinding: An Inner History of the New AmericaRating: 4 out of 5 stars4/5 (45)
- Instrument System Oct 2011Document12 pagesInstrument System Oct 2011Manish GusainNo ratings yet
- Ansi-Hi Pump StandardsDocument2 pagesAnsi-Hi Pump StandardsAndrés Eduardo100% (1)
- Design of CFBC BoilerDocument86 pagesDesign of CFBC Boilerhalder.kalyan88% (8)
- Visual Inspection Checklist For Piping Systems: Connected Mechanical Equipment, Instrumentation, and Pipe Support SystemsDocument2 pagesVisual Inspection Checklist For Piping Systems: Connected Mechanical Equipment, Instrumentation, and Pipe Support SystemsrajuajiNo ratings yet
- O-P20 Broken Glass IncidentDocument3 pagesO-P20 Broken Glass IncidentTinny SumardiNo ratings yet
- 25 HP & 20 HP Solar Pump QuatationDocument5 pages25 HP & 20 HP Solar Pump QuatationAmodNo ratings yet
- Okan Ggcpdebottlenecking Project: Welding Procedure Specification FOR Pipe To FlangeDocument4 pagesOkan Ggcpdebottlenecking Project: Welding Procedure Specification FOR Pipe To FlangeNilesh KabadeNo ratings yet
- Cheaper Raw Materials Sugar and Starch: For Europe'S IndustriesDocument22 pagesCheaper Raw Materials Sugar and Starch: For Europe'S IndustriesTinny SumardiNo ratings yet
- Gerber Test For Fat % in Raw Milk Gerber Test For Fat % in Raw MilkDocument2 pagesGerber Test For Fat % in Raw Milk Gerber Test For Fat % in Raw MilkGerald KwinjoNo ratings yet
- 22 Session, Washington, DC Metro Area, U.S.A., 27 September - 1 October 2004Document11 pages22 Session, Washington, DC Metro Area, U.S.A., 27 September - 1 October 2004Tinny SumardiNo ratings yet
- Fat Tank Glucose Tank Premix Tank I Premix Tank Ii Hot Water Tank High Shear Tank IDocument1 pageFat Tank Glucose Tank Premix Tank I Premix Tank Ii Hot Water Tank High Shear Tank ITinny SumardiNo ratings yet
- TableDocument1 pageTableTinny SumardiNo ratings yet
- Persandingan Standar Edisi 2011 2009Document9 pagesPersandingan Standar Edisi 2011 2009Tinny SumardiNo ratings yet
- Stephanie A. Coronadoa, Graham R. Troutb, Frank R. Dunsheac, Nagendra P. ShahaDocument2 pagesStephanie A. Coronadoa, Graham R. Troutb, Frank R. Dunsheac, Nagendra P. ShahaTinny SumardiNo ratings yet
- Glycemic Control and Insulin Therapy in Sepsis and Critical IllnessDocument1 pageGlycemic Control and Insulin Therapy in Sepsis and Critical IllnessTinny SumardiNo ratings yet
- Dropdown Option:: Please Fill Out This Form For Online AssessmentDocument3 pagesDropdown Option:: Please Fill Out This Form For Online AssessmentTinny SumardiNo ratings yet
- Food Chemistry: Tingting Zhao, Da Som No, Byung Hee Kim, Hugo S. Garcia, Yangha Kim, In-Hwan KimDocument9 pagesFood Chemistry: Tingting Zhao, Da Som No, Byung Hee Kim, Hugo S. Garcia, Yangha Kim, In-Hwan KimTinny SumardiNo ratings yet
- TiketDocument12 pagesTiketTinny SumardiNo ratings yet
- Tugas Kelompok Toksikologi Hasil Perikanan: Keracunan Yang Disebabkan Oleh MolluscaDocument1 pageTugas Kelompok Toksikologi Hasil Perikanan: Keracunan Yang Disebabkan Oleh MolluscaTinny SumardiNo ratings yet
- Ttl/Hcmos Crystal Oscillator: MCO-1510A / MCO-1510B (FULL SIZE)Document1 pageTtl/Hcmos Crystal Oscillator: MCO-1510A / MCO-1510B (FULL SIZE)cutoNo ratings yet
- Establishment of Standard X-Ray Qualities To Be Used in Diagnostic Level at SsdlsDocument50 pagesEstablishment of Standard X-Ray Qualities To Be Used in Diagnostic Level at SsdlsHoomi ShbNo ratings yet
- 6CH02 01 Que 20160610Document24 pages6CH02 01 Que 20160610anon_845676495No ratings yet
- PRIMER - Cost Estimation - Chemical Engineering ProjectsDocument9 pagesPRIMER - Cost Estimation - Chemical Engineering ProjectsMarkoNo ratings yet
- AMF InstructionsDocument4 pagesAMF Instructionspapasp105305No ratings yet
- Hot Bolt Clamp 8pp BrochureDocument8 pagesHot Bolt Clamp 8pp BrochureAlaxxiNo ratings yet
- Daylighting IN Underground Buildings: Ashraf Ali Ibrahim NessimDocument365 pagesDaylighting IN Underground Buildings: Ashraf Ali Ibrahim NessimthabisNo ratings yet
- Product Leaflet - 12V 84ah 1.07kWhDocument2 pagesProduct Leaflet - 12V 84ah 1.07kWhramshukla2001No ratings yet
- Chapter 14 - Shop SafetyDocument11 pagesChapter 14 - Shop Safetymega87_2000No ratings yet
- Reclosers: Form 6 Microprocessor-Based Pole Mount Recloser Control Installation and Operation InstructionsDocument48 pagesReclosers: Form 6 Microprocessor-Based Pole Mount Recloser Control Installation and Operation InstructionsTrịnh Huy ĐảmNo ratings yet
- Quiz For Geoheat PumpsDocument2 pagesQuiz For Geoheat PumpsReach SelfbloodNo ratings yet
- Citizen Ecoodrive B642Document35 pagesCitizen Ecoodrive B642Stefan LaurentiuNo ratings yet
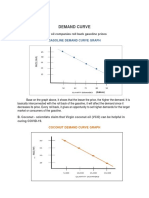
- Demand and Supply CurveDocument3 pagesDemand and Supply CurveJanella Umieh De UngriaNo ratings yet
- Service Manual: LC320EM81Document69 pagesService Manual: LC320EM81wilfredo falconNo ratings yet
- 597 Series PDFDocument52 pages597 Series PDFtystar_21No ratings yet
- SBSBattery VRLA Tubular Gel VRZ Series PDFDocument1 pageSBSBattery VRLA Tubular Gel VRZ Series PDFAleiska Victoria Gómez BetancourthNo ratings yet
- Notes FlamephotometryDocument19 pagesNotes FlamephotometryKaFiAliMirzaNo ratings yet
- 04 - Metode ElektrogravimetriDocument19 pages04 - Metode ElektrogravimetriNurul Aulia HusainNo ratings yet
- TSI Job Aid 1Document8 pagesTSI Job Aid 1mumamaduraiNo ratings yet
- Heat Exchange PDFDocument3 pagesHeat Exchange PDFmunhNo ratings yet
- BCT-UU 48V200Ah LiFePO4 Battery Pack 2021-9-2Document3 pagesBCT-UU 48V200Ah LiFePO4 Battery Pack 2021-9-2Abd AlhadiNo ratings yet
- Atal Aicte Online Faculty Development Programme (FDP)Document6 pagesAtal Aicte Online Faculty Development Programme (FDP)indranildasNo ratings yet
- Topic 1 Numerical Simulation of Cavitating Flows Using OpenfoamDocument7 pagesTopic 1 Numerical Simulation of Cavitating Flows Using OpenfoamKumar ByesNo ratings yet