You might also like
- International Journal of Biological Macromolecules: Manlin Li, Zengqiang Zhang, Ronghua Li, Jim J. Wang, Amjad AliDocument9 pagesInternational Journal of Biological Macromolecules: Manlin Li, Zengqiang Zhang, Ronghua Li, Jim J. Wang, Amjad AliMd. BadrulNo ratings yet
- Adsorption of Micronutrient Metal Ion Onto Struvite To Prepare SL - 2019 - ChemoDocument9 pagesAdsorption of Micronutrient Metal Ion Onto Struvite To Prepare SL - 2019 - ChemoLeynard NatividadNo ratings yet
- Kinetics and Equilibrium Model of PB (Ii) and CD (Ii) Adsorption Onto Tetrakis-Thiomethyl-C-4-Methoxyphenylcalix (4) ResorcinareneDocument8 pagesKinetics and Equilibrium Model of PB (Ii) and CD (Ii) Adsorption Onto Tetrakis-Thiomethyl-C-4-Methoxyphenylcalix (4) ResorcinareneAnonymous ufzCXoidNo ratings yet
- Adsorption of Copper and Chromium by Us CarbonariusDocument5 pagesAdsorption of Copper and Chromium by Us CarbonariusAlejandro Josue Leon MedinaNo ratings yet
- Trace Element Modelling: XIV.1. Why Are We Concerned About Trace Metals ?Document30 pagesTrace Element Modelling: XIV.1. Why Are We Concerned About Trace Metals ?thefunfundaNo ratings yet
- Research Article: Chemical Modifications of Cassava Peel As Adsorbent Material For Metals Ions From WastewaterDocument16 pagesResearch Article: Chemical Modifications of Cassava Peel As Adsorbent Material For Metals Ions From WastewaterLokesh MahadevanNo ratings yet
- Journal of Molecular Catalysis A: ChemicalDocument10 pagesJournal of Molecular Catalysis A: ChemicalAlina IonNo ratings yet
- Hygroscopic Properties of Oxalic Acid and Atmospherically Relevant OxalatesDocument8 pagesHygroscopic Properties of Oxalic Acid and Atmospherically Relevant OxalatesAnisa RachmadaniNo ratings yet
- Baybars Ali Fil, and Mustafa Korkmaz, Cengiz ÖzmetinDocument6 pagesBaybars Ali Fil, and Mustafa Korkmaz, Cengiz ÖzmetinAmmr MahmoodNo ratings yet
- A Chemical AND Thermodynamic Model of Ore Deposition in Hydrothermal SystemsDocument32 pagesA Chemical AND Thermodynamic Model of Ore Deposition in Hydrothermal SystemsDiego JerezNo ratings yet
- Spectrochimica Acta Part A: Molecular and Biomolecular SpectrosDocument9 pagesSpectrochimica Acta Part A: Molecular and Biomolecular SpectrosAakash VNo ratings yet
- Thermal Stability of Naproxen Intercalated in Layered Double HydroxideDocument8 pagesThermal Stability of Naproxen Intercalated in Layered Double Hydroxidejaimeo_07No ratings yet
- J Cej 2010 12 029Document8 pagesJ Cej 2010 12 029Muhammad Furqan FayazNo ratings yet
- Madam Shazia PaperDocument14 pagesMadam Shazia PaperpervaizhejNo ratings yet
- Extraccion Secuencial-Tessier 1979Document8 pagesExtraccion Secuencial-Tessier 1979Alex Rhdz100% (1)
- Pyrite Behaviour in A Tailings Pond: C. Garcı A, A. Ballester, F. Gonza Lez, M.L. Bla ZquezDocument12 pagesPyrite Behaviour in A Tailings Pond: C. Garcı A, A. Ballester, F. Gonza Lez, M.L. Bla Zquezosvaldohumberto1974No ratings yet
- Accepted Manuscript: International Journal of Biological MacromoleculesDocument38 pagesAccepted Manuscript: International Journal of Biological MacromoleculesAzhar AbbasNo ratings yet
- Literature Review of Biosorption of Heavy MetalsDocument5 pagesLiterature Review of Biosorption of Heavy Metalsafdttricd100% (1)
- Ashraf, Mahmood, Dan Wajid (2011) PDFDocument9 pagesAshraf, Mahmood, Dan Wajid (2011) PDFMiranti PuspitasariNo ratings yet
- Synthesis of New Schiff Base From Natural Products For Remediation of Water Pollution With Heavy Metals in Industrial AreasDocument10 pagesSynthesis of New Schiff Base From Natural Products For Remediation of Water Pollution With Heavy Metals in Industrial AreasChern YuanNo ratings yet
- Desalination: Yian Zheng, Shuibo Hua, Aiqin WangDocument6 pagesDesalination: Yian Zheng, Shuibo Hua, Aiqin WangAnonymous 9XI54PvKPNo ratings yet
- Research ArticleDocument17 pagesResearch ArticleSoussou PerlaNo ratings yet
- Petit Et Al 2009Document23 pagesPetit Et Al 2009LudNo ratings yet
- Substrate Inhibition: Oxidation of D - Sorbitol and D - Mannitol by Potassium Periodate in Alkaline MediumDocument5 pagesSubstrate Inhibition: Oxidation of D - Sorbitol and D - Mannitol by Potassium Periodate in Alkaline MediumHeidi HughesNo ratings yet
- Wheat StrawDocument11 pagesWheat StrawwaleeNo ratings yet
- Adsorption Study of Anionic Reactive Dye From Aqueous Solution To Mg-Fe-CO Layered Double Hydroxide (LDH)Document7 pagesAdsorption Study of Anionic Reactive Dye From Aqueous Solution To Mg-Fe-CO Layered Double Hydroxide (LDH)Suila KimberlyNo ratings yet
- Adsorp Cu Sekam PadiDocument7 pagesAdsorp Cu Sekam PadiDinda JuwitaNo ratings yet
- Algal Sulfur Compound Release in Response to Light RegimesDocument8 pagesAlgal Sulfur Compound Release in Response to Light RegimesErika M. Sánchez MaquiNo ratings yet
- 1 s2.0 S0045653599003021 Main PDFDocument7 pages1 s2.0 S0045653599003021 Main PDFyuryandrea01No ratings yet
- Catalysis Today: Balázs Réti, Gabriella Ilona Kiss, Tamás Gyulavári, Kornelia Baan, Klara Magyari, Klara HernadiDocument9 pagesCatalysis Today: Balázs Réti, Gabriella Ilona Kiss, Tamás Gyulavári, Kornelia Baan, Klara Magyari, Klara HernadiŞebnem Gül İlarslanNo ratings yet
- Determination Foods eDocument7 pagesDetermination Foods eRoberta MatosNo ratings yet
- A Solute Transport Model For The Acid Leaching of Copper in Soil ColumnsDocument9 pagesA Solute Transport Model For The Acid Leaching of Copper in Soil Columnsalexis diazNo ratings yet
- Catalysts 11 00713Document17 pagesCatalysts 11 00713Quoc LongNo ratings yet
- Nadeem Sir1Document6 pagesNadeem Sir1baquir_aligNo ratings yet
- Syllabus Chemistry (UG Courses) Admitted Batch 2008 - 2009Document33 pagesSyllabus Chemistry (UG Courses) Admitted Batch 2008 - 2009ArunNo ratings yet
- Articolo A.torreggianiDocument8 pagesArticolo A.torreggianirosarioNo ratings yet
- Competition and Complexation of Heavy Metal Ions and Humic Acid On Zeolitic MCM-22 and Activated CarbonDocument8 pagesCompetition and Complexation of Heavy Metal Ions and Humic Acid On Zeolitic MCM-22 and Activated CarbonMurat AkdoğanNo ratings yet
- Characterization of Gas Hydrates With PXRD, DSC, NMR, and RamanDocument10 pagesCharacterization of Gas Hydrates With PXRD, DSC, NMR, and RamanDiana GarciaNo ratings yet
- Trace Analysis Principles SummaryDocument8 pagesTrace Analysis Principles SummarySon TaifaNo ratings yet
- Liang 2010Document7 pagesLiang 2010GERSON JOEL ORTEGA MORALESNo ratings yet
- Adsorption of Nickel in Water by Brown Algae: Laminaria Japonica and Undaria PinnatifidaDocument3 pagesAdsorption of Nickel in Water by Brown Algae: Laminaria Japonica and Undaria PinnatifidaRakesh SHNo ratings yet
- Chemosphere: Erik A. Rodríguez-Morales, Eduardo Rodríguez de San Miguel, Jose Fina de GyvesDocument11 pagesChemosphere: Erik A. Rodríguez-Morales, Eduardo Rodríguez de San Miguel, Jose Fina de GyvesrafumiNo ratings yet
- Adsorption of Congo Red Dye Using Mg-Al Layered Double HydroxideDocument6 pagesAdsorption of Congo Red Dye Using Mg-Al Layered Double HydroxideLucas Ferreira LozNo ratings yet
- Pagination BIOMAC 21357Document10 pagesPagination BIOMAC 21357Yahya RajputNo ratings yet
- Ziegler KJ 2003bDocument6 pagesZiegler KJ 2003bRiyasath ReihanNo ratings yet
- Modelling of Copper Adsorption FinalDocument21 pagesModelling of Copper Adsorption FinalaadhyaNo ratings yet
- Modelling of Copper Adsorption FinalDocument21 pagesModelling of Copper Adsorption FinalaadhyaNo ratings yet
- Ls Con Rayos Gamma CaractDocument7 pagesLs Con Rayos Gamma CaractVerónica Sáez JiménezNo ratings yet
- Water Resources Research - February 1995 - Chapelle - Deducing The Distribution of Terminal Electron Accepting Processes inDocument13 pagesWater Resources Research - February 1995 - Chapelle - Deducing The Distribution of Terminal Electron Accepting Processes inIsabella GagoNo ratings yet
- 12.study On The Removal of PB (II) Ions From Aqueous Solution Using Chemically Modified Corn Cob. (Siti Raihan Zakaria) PP 74-81Document8 pages12.study On The Removal of PB (II) Ions From Aqueous Solution Using Chemically Modified Corn Cob. (Siti Raihan Zakaria) PP 74-81upenapahangNo ratings yet
- On The Applicability of DLVO Theory To The Prediction of Clay Colloids Stability - MissanaDocument7 pagesOn The Applicability of DLVO Theory To The Prediction of Clay Colloids Stability - MissanalearningboxNo ratings yet
- 4088 20368 4 PB PDFDocument12 pages4088 20368 4 PB PDFVersel ArendaNo ratings yet
- LDH SpectraDocument7 pagesLDH SpectraShamsheer KhanNo ratings yet
- Effectiveness of Sunflower Seed Husk Biochar For Removing Cu 2+ in Waste WaterDocument11 pagesEffectiveness of Sunflower Seed Husk Biochar For Removing Cu 2+ in Waste WaterMuhamad SuharNo ratings yet
- Chemical Physics LettersDocument6 pagesChemical Physics LettersBrenda Valeria López lopezNo ratings yet
- Synthesis and Evaluation Catalytic Efficiency of Perovskite-Type Oxide Nanopowders in Removal of Bromocresol Purple From Aqueous SolutionDocument12 pagesSynthesis and Evaluation Catalytic Efficiency of Perovskite-Type Oxide Nanopowders in Removal of Bromocresol Purple From Aqueous SolutionAmin MojiriNo ratings yet
- Heavy metal removal using aquatic plantDocument4 pagesHeavy metal removal using aquatic plantRamona DanyNo ratings yet
- Tessier-1979-Sequential Extraction Procedure For The SpeciatDocument10 pagesTessier-1979-Sequential Extraction Procedure For The SpeciatRama NamikazeNo ratings yet
- Geofluids: Developments in Microthermometry, Spectroscopy, Thermodynamics, and Stable IsotopesFrom EverandGeofluids: Developments in Microthermometry, Spectroscopy, Thermodynamics, and Stable IsotopesNo ratings yet
- The Determination of Carboxylic Functional Groups: Monographs in Organic Functional Group AnalysisFrom EverandThe Determination of Carboxylic Functional Groups: Monographs in Organic Functional Group AnalysisNo ratings yet
- CarbohydratesDocument1 pageCarbohydratesFazreen DzulkafliNo ratings yet
- Cadmium (CD) and Nickel (Ni) Distribution On Size-Fractioned Soil Humic Substance (SHS)Document11 pagesCadmium (CD) and Nickel (Ni) Distribution On Size-Fractioned Soil Humic Substance (SHS)Fazreen DzulkafliNo ratings yet
- OTGA INOS Marine GIS Tools ICZM Flyer - Dec2018Document1 pageOTGA INOS Marine GIS Tools ICZM Flyer - Dec2018Fazreen DzulkafliNo ratings yet
- PREDICTDocument3 pagesPREDICTFazreen DzulkafliNo ratings yet
- Is The Traditional Alkali Extraction Method ValidDocument16 pagesIs The Traditional Alkali Extraction Method ValidFazreen DzulkafliNo ratings yet
- Lab Manual BiochemDocument8 pagesLab Manual BiochemFazreen DzulkafliNo ratings yet
- CH 3 Proteins Student PrintDocument23 pagesCH 3 Proteins Student PrintFazreen DzulkafliNo ratings yet
- Environmental audit of a distillery industryDocument5 pagesEnvironmental audit of a distillery industryFazreen DzulkafliNo ratings yet
- 5-Ocean Thermal Energy Conversion The Promise of A Clean FutureDocument4 pages5-Ocean Thermal Energy Conversion The Promise of A Clean FutureFazreen DzulkafliNo ratings yet
- Biochemistry Test 1Document2 pagesBiochemistry Test 1Fazreen DzulkafliNo ratings yet
- Stages 2011 2012 2013: Gantt Chart of Research ActivitiesDocument1 pageStages 2011 2012 2013: Gantt Chart of Research ActivitiesFazreen DzulkafliNo ratings yet
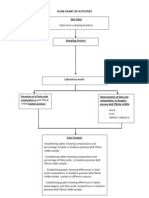
- Flow Chart of ActivitiesDocument1 pageFlow Chart of ActivitiesFazreen DzulkafliNo ratings yet
- Dolni VestoniceDocument34 pagesDolni VestoniceOlha PodufalovaNo ratings yet
- Excel Solver Optimization ReportDocument9 pagesExcel Solver Optimization ReportMy Duyen NguyenNo ratings yet
- Differential Pulse Code ModulationDocument12 pagesDifferential Pulse Code ModulationNarasimhareddy MmkNo ratings yet
- HVAC Master Validation PlanDocument51 pagesHVAC Master Validation Plannavas197293% (30)
- Statistical Quality Control, 7th Edition by Douglas C. Montgomery. 1Document76 pagesStatistical Quality Control, 7th Edition by Douglas C. Montgomery. 1omerfaruk200141No ratings yet
- Pasadena Nursery Roses Inventory ReportDocument2 pagesPasadena Nursery Roses Inventory ReportHeng SrunNo ratings yet
- Certification Presently EnrolledDocument15 pagesCertification Presently EnrolledMaymay AuauNo ratings yet
- Weone ProfileDocument10 pagesWeone ProfileOmair FarooqNo ratings yet
- Eye Bags ReliefDocument27 pagesEye Bags ReliefNatsu DragneelNo ratings yet
- Stroboscopy For Benign Laryngeal Pathology in Evidence Based Health CareDocument5 pagesStroboscopy For Benign Laryngeal Pathology in Evidence Based Health CareDoina RusuNo ratings yet
- Sinclair User 1 Apr 1982Document68 pagesSinclair User 1 Apr 1982JasonWhite99No ratings yet
- 4 Influencing Factors of Learners Career Choice Parents Choice Vs Personal DescisionDocument24 pages4 Influencing Factors of Learners Career Choice Parents Choice Vs Personal Descisionmatteo mamaloNo ratings yet
- Production of Sodium Chlorite PDFDocument13 pagesProduction of Sodium Chlorite PDFangelofgloryNo ratings yet
- Cableado de TermocuplasDocument3 pagesCableado de TermocuplasRUBEN DARIO BUCHELLYNo ratings yet
- Oxgen Sensor Cat WEBDocument184 pagesOxgen Sensor Cat WEBBuddy Davis100% (2)
- Flexible Regression and Smoothing - Using GAMLSS in RDocument572 pagesFlexible Regression and Smoothing - Using GAMLSS in RDavid50% (2)
- Pfr140 User ManualDocument4 pagesPfr140 User ManualOanh NguyenNo ratings yet
- MA1201 Calculus and Basic Linear Algebra II Solution of Problem Set 4Document10 pagesMA1201 Calculus and Basic Linear Algebra II Solution of Problem Set 4Sit LucasNo ratings yet
- Strategies For StartupDocument16 pagesStrategies For StartupRoshankumar BalasubramanianNo ratings yet
- 2020 Global Finance Business Management Analyst Program - IIMDocument4 pages2020 Global Finance Business Management Analyst Program - IIMrishabhaaaNo ratings yet
- 3d Control Sphere Edge and Face StudyDocument4 pages3d Control Sphere Edge and Face Studydjbroussard100% (2)
- Training Customer CareDocument6 pagesTraining Customer Careyahya sabilNo ratings yet
- Problem Set SolutionsDocument16 pagesProblem Set SolutionsKunal SharmaNo ratings yet
- Pipeline Welding SpecificationDocument15 pagesPipeline Welding Specificationaslam.ambNo ratings yet
- What Is A Problem?: Method + Answer SolutionDocument17 pagesWhat Is A Problem?: Method + Answer SolutionShailaMae VillegasNo ratings yet
- Dermatology Study Guide 2023-IvDocument7 pagesDermatology Study Guide 2023-IvUnknown ManNo ratings yet
- U2 All That You Can't Leave BehindDocument82 pagesU2 All That You Can't Leave BehindFranck UrsiniNo ratings yet
- Evaluative Research DesignDocument17 pagesEvaluative Research DesignMary Grace BroquezaNo ratings yet
- Key Fact Sheet (HBL FreedomAccount) - July 2019 PDFDocument1 pageKey Fact Sheet (HBL FreedomAccount) - July 2019 PDFBaD cHaUhDrYNo ratings yet