You might also like
- Shoe Dog: A Memoir by the Creator of NikeFrom EverandShoe Dog: A Memoir by the Creator of NikeRating: 4.5 out of 5 stars4.5/5 (537)
- Grit: The Power of Passion and PerseveranceFrom EverandGrit: The Power of Passion and PerseveranceRating: 4 out of 5 stars4/5 (587)
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceFrom EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceRating: 4 out of 5 stars4/5 (890)
- The Yellow House: A Memoir (2019 National Book Award Winner)From EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Rating: 4 out of 5 stars4/5 (98)
- The Little Book of Hygge: Danish Secrets to Happy LivingFrom EverandThe Little Book of Hygge: Danish Secrets to Happy LivingRating: 3.5 out of 5 stars3.5/5 (399)
- On Fire: The (Burning) Case for a Green New DealFrom EverandOn Fire: The (Burning) Case for a Green New DealRating: 4 out of 5 stars4/5 (73)
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeFrom EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeRating: 4 out of 5 stars4/5 (5794)
- Never Split the Difference: Negotiating As If Your Life Depended On ItFrom EverandNever Split the Difference: Negotiating As If Your Life Depended On ItRating: 4.5 out of 5 stars4.5/5 (838)
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureFrom EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureRating: 4.5 out of 5 stars4.5/5 (474)
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryFrom EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryRating: 3.5 out of 5 stars3.5/5 (231)
- The Emperor of All Maladies: A Biography of CancerFrom EverandThe Emperor of All Maladies: A Biography of CancerRating: 4.5 out of 5 stars4.5/5 (271)
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreFrom EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreRating: 4 out of 5 stars4/5 (1090)
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyFrom EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyRating: 3.5 out of 5 stars3.5/5 (2219)
- Team of Rivals: The Political Genius of Abraham LincolnFrom EverandTeam of Rivals: The Political Genius of Abraham LincolnRating: 4.5 out of 5 stars4.5/5 (234)
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersFrom EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersRating: 4.5 out of 5 stars4.5/5 (344)
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaFrom EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaRating: 4.5 out of 5 stars4.5/5 (265)
- The Unwinding: An Inner History of the New AmericaFrom EverandThe Unwinding: An Inner History of the New AmericaRating: 4 out of 5 stars4/5 (45)
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)From EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Rating: 4.5 out of 5 stars4.5/5 (119)
- Her Body and Other Parties: StoriesFrom EverandHer Body and Other Parties: StoriesRating: 4 out of 5 stars4/5 (821)
- Trends of Measles in Nigeria A Systematic ReviewDocument7 pagesTrends of Measles in Nigeria A Systematic ReviewRizki Nandasari SulbahriNo ratings yet
- Lenv62012 Exam WDocument24 pagesLenv62012 Exam Wcarlapaulavicente3774No ratings yet
- Writing Practice For Toefl IbtDocument4 pagesWriting Practice For Toefl IbtsasiarchieNo ratings yet
- Role of Atypical Pathogens in The Etiology of Community-Acquired PneumoniaDocument10 pagesRole of Atypical Pathogens in The Etiology of Community-Acquired PneumoniaJaya Semara PutraNo ratings yet
- Womens Health Core CurriculumDocument152 pagesWomens Health Core Curriculumemaleth00100% (2)
- Communicable Disease Nursing Part II Diseases (1) 2Document21 pagesCommunicable Disease Nursing Part II Diseases (1) 2MK LiNo ratings yet
- Morphology of Moulds/ Molds A. ThallusDocument3 pagesMorphology of Moulds/ Molds A. ThallusJane LappaoNo ratings yet
- DLP 3Q - Pathogens - SampleDocument2 pagesDLP 3Q - Pathogens - SampleJords AureaNo ratings yet
- What Is BrucellosisDocument2 pagesWhat Is BrucellosisSumbul RafatNo ratings yet
- The Importance of Vaccination During A PandemicDocument2 pagesThe Importance of Vaccination During A PandemicSalsabilla KusumaningrumNo ratings yet
- EBCR Hepatologi - Edel Herbitya 1706098814Document34 pagesEBCR Hepatologi - Edel Herbitya 1706098814Fita FitriantiNo ratings yet
- Biological HazaDocument3 pagesBiological HazaVladimere Censon ReyesNo ratings yet
- L95 Expert PanelDocument3 pagesL95 Expert PanelKING 5 NewsNo ratings yet
- Enloe infectionPreventionAndControl QuizDocument3 pagesEnloe infectionPreventionAndControl QuizsarahhNo ratings yet
- Kenya-Comprehensive Multi-Year Plan For 2011-2015 - Year UnknownDocument76 pagesKenya-Comprehensive Multi-Year Plan For 2011-2015 - Year UnknownSalman MajidNo ratings yet
- Edited Monkeypox Facts 2022Document93 pagesEdited Monkeypox Facts 2022anghelmiguel07262016No ratings yet
- Barangay Annual Budget Youth Investment Plan (ABYIP) for 2019Document4 pagesBarangay Annual Budget Youth Investment Plan (ABYIP) for 2019Allen GomintongNo ratings yet
- Malaria Is A Mosquito-Borne Infectious Disease of Humans and OtherDocument3 pagesMalaria Is A Mosquito-Borne Infectious Disease of Humans and Otherkiran PoudelNo ratings yet
- Cap Color GuideDocument2 pagesCap Color GuideBryan NicollNo ratings yet
- Chapter Overview: 8: FungiDocument17 pagesChapter Overview: 8: FungiBianca ElbrechtNo ratings yet
- Managing Pain After AppendectomyDocument2 pagesManaging Pain After AppendectomyChatoh SanaoNo ratings yet
- Saudi Journal of Biological SciencesDocument7 pagesSaudi Journal of Biological Sciencesgood doctorNo ratings yet
- ZCM 822Document59 pagesZCM 822Ema GuillotineNo ratings yet
- Cot 1 in Health 8 2021Document4 pagesCot 1 in Health 8 2021Anne LacsamanaNo ratings yet
- Model PaperDocument19 pagesModel PaperShravanthi KonaNo ratings yet
- Sultamicillin, Hydroxyurea, Paracetamol, NystatinDocument4 pagesSultamicillin, Hydroxyurea, Paracetamol, NystatinLeodel Tolentino Barrio100% (1)
- Acute Hematogenous Osteomyelitis in ChildrenDocument63 pagesAcute Hematogenous Osteomyelitis in ChildrenCati Moraru100% (1)
- Pneumonia PresentationDocument23 pagesPneumonia Presentationapi-546694141No ratings yet
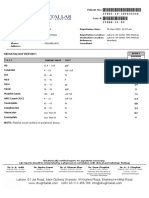
- Hematology Report:: Lahore: 07 Jail Road, Main Gulberg - Karachi: Al Khaleej Plaza, Shaheed-e-Millat RoadDocument2 pagesHematology Report:: Lahore: 07 Jail Road, Main Gulberg - Karachi: Al Khaleej Plaza, Shaheed-e-Millat RoadAliNo ratings yet
- Verorab Drug StudyDocument2 pagesVerorab Drug StudyHanna SeNo ratings yet