You might also like
- Removing The Mystery of Entropy and Thermodynamics - Part I: Related ArticlesDocument5 pagesRemoving The Mystery of Entropy and Thermodynamics - Part I: Related ArticlesPeterNo ratings yet
- “Foundations to Flight: Mastering Physics from Curiosity to Confidence: Cipher 4”: “Foundations to Flight: Mastering Physics from Curiosity to Confidence, #4From Everand“Foundations to Flight: Mastering Physics from Curiosity to Confidence: Cipher 4”: “Foundations to Flight: Mastering Physics from Curiosity to Confidence, #4No ratings yet
- Book Summary: A. The Nature of Energy and Types of EnergyDocument9 pagesBook Summary: A. The Nature of Energy and Types of EnergyFildzahNo ratings yet
- First Law of Thermodynamics - Reversible and Irreversible ProcessDocument8 pagesFirst Law of Thermodynamics - Reversible and Irreversible ProcessLucas FagundesNo ratings yet
- Laws of ThermodynamicsDocument4 pagesLaws of ThermodynamicsAyan BarbonNo ratings yet
- Internal Energy and The Ideal GasDocument9 pagesInternal Energy and The Ideal GasfagroupandahmadsonsNo ratings yet
- UntitledDocument16 pagesUntitledapi-233404189100% (1)
- Fundamentals of Chemical ThermodynamicsDocument57 pagesFundamentals of Chemical ThermodynamicstNo ratings yet
- Me 291 Introduction To Mechechanical EngineeringDocument11 pagesMe 291 Introduction To Mechechanical EngineeringDaniel Deng KuolNo ratings yet
- 5 ThermodynamicsDocument16 pages5 Thermodynamicsabmusic711No ratings yet
- 22 - MSN 212 - 2013 - Konduksi-Konveksi Dan RadiasiDocument4 pages22 - MSN 212 - 2013 - Konduksi-Konveksi Dan RadiasiReza Dwi PutraNo ratings yet
- Unit IV ThermodynamicsDocument7 pagesUnit IV ThermodynamicsAyush KatiyarNo ratings yet
- QuestionsDocument8 pagesQuestionsAntonioNo ratings yet
- Work and Heat Concepts in ThermodynamicsDocument13 pagesWork and Heat Concepts in ThermodynamicsAkol MajookNo ratings yet
- BCH 2202 Chemical Thermodynamics ModuleDocument84 pagesBCH 2202 Chemical Thermodynamics Modulefrancis100% (5)
- Energy, Work and Heat: Lesson 4Document5 pagesEnergy, Work and Heat: Lesson 4Jhelyne FloresNo ratings yet
- Thermodynamics WrittenWork1Document2 pagesThermodynamics WrittenWork1MikoNo ratings yet
- 1ST Law of ThermodynamicsDocument7 pages1ST Law of ThermodynamicsKen BorjaNo ratings yet
- CHAPTER 3: First Law of Thermodynamics: 3.1 Concept of Internal EnergyDocument18 pagesCHAPTER 3: First Law of Thermodynamics: 3.1 Concept of Internal EnergyMangesh UgrankarNo ratings yet
- Energetics of Chemical ReactionsDocument58 pagesEnergetics of Chemical ReactionsBichitra GautamNo ratings yet
- Laporan Suwandy Effendy Unit 1 (FINISH)Document39 pagesLaporan Suwandy Effendy Unit 1 (FINISH)maharani fauzia annurNo ratings yet
- Fundamentals of Chemical ThermodynamicsDocument57 pagesFundamentals of Chemical ThermodynamicstNo ratings yet
- Thermodynamics: Large Scale Response Kinetic Theory Thermodynamics, HeatDocument30 pagesThermodynamics: Large Scale Response Kinetic Theory Thermodynamics, HeatKritika KapoorNo ratings yet
- Thermochemistry: Energy Flow and Chemical ChangeDocument59 pagesThermochemistry: Energy Flow and Chemical ChangeMuhammad Zahir100% (1)
- Molecules. in General, Thermal Energy Can Be Calculated From Temperature Measurements. TheDocument6 pagesMolecules. in General, Thermal Energy Can Be Calculated From Temperature Measurements. Thesari wahyuniNo ratings yet
- Physics (H.R.K) Chapter 25: HeatDocument10 pagesPhysics (H.R.K) Chapter 25: HeatSaqlain YousufNo ratings yet
- Energy, Heat, and WorkDocument8 pagesEnergy, Heat, and WorkCristina Tubig CalarianNo ratings yet
- The Ideal Gas Law and The Kinetic Theory of GasesDocument17 pagesThe Ideal Gas Law and The Kinetic Theory of GasesapexrapperNo ratings yet
- CO3 ThermodynamicsDocument72 pagesCO3 ThermodynamicsRonna IturaldeNo ratings yet
- CHE 110 - 05 - Principles of Chemical Reactivity - Energy and Chemical Reactions - ThermochemistryDocument58 pagesCHE 110 - 05 - Principles of Chemical Reactivity - Energy and Chemical Reactions - ThermochemistryJoachim MotoNo ratings yet
- Statistical Mechanics: Alejandro L. GarciaDocument133 pagesStatistical Mechanics: Alejandro L. GarciaAnonymous YEDUXNNo ratings yet
- Chapter 6 IM Chang 11eDocument8 pagesChapter 6 IM Chang 11eSelma MeloNo ratings yet
- Introduction To ThermodynamicsDocument131 pagesIntroduction To Thermodynamicsmanosmagicas7340No ratings yet
- Thermodynamics 1 LectureDocument57 pagesThermodynamics 1 LecturefabyunaaaNo ratings yet
- Thermodynamics 1 LectureDocument57 pagesThermodynamics 1 LecturefabyunaaaNo ratings yet
- CHEM 1050 - General Chemistry II: Introduction To THERMODYNAMICSDocument64 pagesCHEM 1050 - General Chemistry II: Introduction To THERMODYNAMICSMarikNo ratings yet
- Topic 3: Thermal Physics: Revision of Everything in Topic 3 of The IB SyllabusDocument6 pagesTopic 3: Thermal Physics: Revision of Everything in Topic 3 of The IB SyllabusUnduh BerkasNo ratings yet
- ThermodynamicsDocument21 pagesThermodynamicsXenon ClassesNo ratings yet
- Kech106 PDFDocument31 pagesKech106 PDFVivek JainNo ratings yet
- Chapter 4 First Law1Document74 pagesChapter 4 First Law1Mihir Kumar MechNo ratings yet
- Mole: 1 Mole of A Substance Contains Avogadro's Number (N 6.02E23)Document53 pagesMole: 1 Mole of A Substance Contains Avogadro's Number (N 6.02E23)Juan Carlos Gonzalez LNo ratings yet
- The First Law and Other Basic Concepts (Part 1)Document23 pagesThe First Law and Other Basic Concepts (Part 1)Ragil BudiartoNo ratings yet
- Chemistry 11th Edition Chang Solutions ManualDocument9 pagesChemistry 11th Edition Chang Solutions Manualcleopatrasang611py100% (33)
- Ebook Chemistry 11Th Edition Chang Solutions Manual Full Chapter PDFDocument30 pagesEbook Chemistry 11Th Edition Chang Solutions Manual Full Chapter PDFJaniceMarqueznxed100% (13)
- Thermal Engineering SumaryDocument3 pagesThermal Engineering SumaryL-IONSNo ratings yet
- Thermodynamics: Specific Learning Objective at The End of The Session The Student Should Be Able To ExplainDocument67 pagesThermodynamics: Specific Learning Objective at The End of The Session The Student Should Be Able To ExplainfitriNo ratings yet
- GP1 - Q2 - Week 7Document6 pagesGP1 - Q2 - Week 7Shekaina Faith Cuizon Lozada100% (1)
- 11 Chemistry Notes ch06 Thermodynamics PDFDocument4 pages11 Chemistry Notes ch06 Thermodynamics PDFRangbaaz DA FIRENZENo ratings yet
- CBSE Class 11 Chemistry Revision Notes Thermodynamics: Material Downloaded From - 1 / 4Document4 pagesCBSE Class 11 Chemistry Revision Notes Thermodynamics: Material Downloaded From - 1 / 4JwalantNo ratings yet
- 340 Sample ChapterDocument49 pages340 Sample ChapterRajbir SinghNo ratings yet
- Thermo ReviewDocument8 pagesThermo ReviewJosh CohenNo ratings yet
- Introduction To ThermodynamicsDocument17 pagesIntroduction To Thermodynamicsveronica NgunziNo ratings yet
- EOS Residuales Thermodynamics & Chemicals Kinetics (ChBC-34)Document292 pagesEOS Residuales Thermodynamics & Chemicals Kinetics (ChBC-34)swerty619No ratings yet
- FALLSEM2020-21 MEE1003 TH VL2020210103023 Reference Material I 31-Jul-2020 First Law of Thermodynamics - IIDocument52 pagesFALLSEM2020-21 MEE1003 TH VL2020210103023 Reference Material I 31-Jul-2020 First Law of Thermodynamics - IIRahul rajelliNo ratings yet
- College Physics 2e-WEB 7zesafu RemovedDocument4 pagesCollege Physics 2e-WEB 7zesafu Removednantespirlo123No ratings yet
- Classical Thermodynamics: The First LawDocument18 pagesClassical Thermodynamics: The First LawMano AnaNo ratings yet
- Classical Thermodynamics: The First LawDocument18 pagesClassical Thermodynamics: The First Lawnoor kareemNo ratings yet
- Thermochemistry or Junior IntermideateDocument48 pagesThermochemistry or Junior IntermideateEvs GoudNo ratings yet
- Liquitex Soft Body BookletDocument12 pagesLiquitex Soft Body Booklethello belloNo ratings yet
- Chief Complaint: History TakingDocument9 pagesChief Complaint: History TakingMohamad ZulfikarNo ratings yet
- Extract The .Msi FilesDocument2 pagesExtract The .Msi FilesvladimirNo ratings yet
- A2Document4 pagesA2Akshay KumarNo ratings yet
- Generalized Class of Sakaguchi Functions in Conic Region: Saritha. G. P, Fuad. S. Al Sarari, S. LathaDocument5 pagesGeneralized Class of Sakaguchi Functions in Conic Region: Saritha. G. P, Fuad. S. Al Sarari, S. LathaerpublicationNo ratings yet
- Just in Time and TQMDocument8 pagesJust in Time and TQMBhramadhathNo ratings yet
- Inventions Over The Last 100 YearsDocument3 pagesInventions Over The Last 100 YearsHombreMorado GamerYTNo ratings yet
- Benefits and Limitations of BEPDocument2 pagesBenefits and Limitations of BEPAnishaAppuNo ratings yet
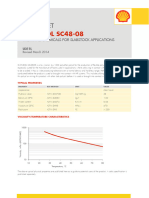
- Caradol sc48 08Document2 pagesCaradol sc48 08GİZEM DEMİRNo ratings yet
- Crafer. The Apocriticus of Macarius Magnes (S.P.C.K. Edition) - 1919.Document188 pagesCrafer. The Apocriticus of Macarius Magnes (S.P.C.K. Edition) - 1919.Patrologia Latina, Graeca et OrientalisNo ratings yet
- Lect2 - 1151 - Grillage AnalysisDocument31 pagesLect2 - 1151 - Grillage AnalysisCheong100% (1)
- Rishika Reddy Art Integrated ActivityDocument11 pagesRishika Reddy Art Integrated ActivityRishika ReddyNo ratings yet
- The Time Machine Was First Published in 1984 As A Story Under The Name The Time Traveller in The National ObserverDocument1 pageThe Time Machine Was First Published in 1984 As A Story Under The Name The Time Traveller in The National ObservermarceNo ratings yet
- The Comma Rules Conversion 15 SlidesDocument15 pagesThe Comma Rules Conversion 15 SlidesToh Choon HongNo ratings yet
- John Wren-Lewis - NDEDocument7 pagesJohn Wren-Lewis - NDEpointandspaceNo ratings yet
- Paul Spicker - The Welfare State A General TheoryDocument162 pagesPaul Spicker - The Welfare State A General TheoryTista ArumNo ratings yet
- 300u Specs Diodo 300 Amps. 25 Dolares RadiosurtidoraDocument6 pages300u Specs Diodo 300 Amps. 25 Dolares RadiosurtidorarepelindNo ratings yet
- Calculus For The Life Sciences 2nd Edition Greenwell Solutions ManualDocument26 pagesCalculus For The Life Sciences 2nd Edition Greenwell Solutions ManualSharonPerezozqy100% (56)
- 4B - Urp - Shavya's FarmDocument22 pages4B - Urp - Shavya's FarmSnehansh KishoreNo ratings yet
- DS Important QuestionsDocument15 pagesDS Important QuestionsLavanya JNo ratings yet
- Clint Freeman ResumeDocument2 pagesClint Freeman ResumeClint Tiberius FreemanNo ratings yet
- Ancient Sumer Flip BookDocument9 pagesAncient Sumer Flip Bookapi-198624210No ratings yet
- Artificial Intelligence Practical 1Document5 pagesArtificial Intelligence Practical 1sadani1989No ratings yet
- Coal Bottom Ash As Sand Replacement in ConcreteDocument9 pagesCoal Bottom Ash As Sand Replacement in ConcretexxqNo ratings yet
- BÀI TẬP LESSON 7. CÂU BỊ ĐỘNG 1Document4 pagesBÀI TẬP LESSON 7. CÂU BỊ ĐỘNG 1Yến Vy TrầnNo ratings yet
- Persuasive Speech 2016 - Whole Person ParadigmDocument4 pagesPersuasive Speech 2016 - Whole Person Paradigmapi-311375616No ratings yet
- Taoist Master Zhang 张天师Document9 pagesTaoist Master Zhang 张天师QiLeGeGe 麒樂格格100% (2)
- Twin PregnancyDocument73 pagesTwin Pregnancykrishna mandalNo ratings yet
- Aribah Ahmed CertificateDocument2 pagesAribah Ahmed CertificateBahadur AliNo ratings yet
- Unit 2 - Industrial Engineering & Ergonomics - WWW - Rgpvnotes.inDocument15 pagesUnit 2 - Industrial Engineering & Ergonomics - WWW - Rgpvnotes.inSACHIN HANAGALNo ratings yet
- The Secret Teachings Of All Ages: AN ENCYCLOPEDIC OUTLINE OF MASONIC, HERMETIC, QABBALISTIC AND ROSICRUCIAN SYMBOLICAL PHILOSOPHYFrom EverandThe Secret Teachings Of All Ages: AN ENCYCLOPEDIC OUTLINE OF MASONIC, HERMETIC, QABBALISTIC AND ROSICRUCIAN SYMBOLICAL PHILOSOPHYRating: 4.5 out of 5 stars4.5/5 (4)
- The Stoic Mindset: Living the Ten Principles of StoicismFrom EverandThe Stoic Mindset: Living the Ten Principles of StoicismRating: 5 out of 5 stars5/5 (6)
- Summary: The Laws of Human Nature: by Robert Greene: Key Takeaways, Summary & AnalysisFrom EverandSummary: The Laws of Human Nature: by Robert Greene: Key Takeaways, Summary & AnalysisRating: 4.5 out of 5 stars4.5/5 (30)
- Stoicism: How to Use Stoic Philosophy to Find Inner Peace and HappinessFrom EverandStoicism: How to Use Stoic Philosophy to Find Inner Peace and HappinessRating: 4.5 out of 5 stars4.5/5 (84)
- 12 Rules for Life by Jordan B. Peterson - Book Summary: An Antidote to ChaosFrom Everand12 Rules for Life by Jordan B. Peterson - Book Summary: An Antidote to ChaosRating: 4.5 out of 5 stars4.5/5 (207)
- How States Think: The Rationality of Foreign PolicyFrom EverandHow States Think: The Rationality of Foreign PolicyRating: 5 out of 5 stars5/5 (7)
- Stoicism The Art of Happiness: How the Stoic Philosophy Works, Living a Good Life, Finding Calm and Managing Your Emotions in a Turbulent World. New VersionFrom EverandStoicism The Art of Happiness: How the Stoic Philosophy Works, Living a Good Life, Finding Calm and Managing Your Emotions in a Turbulent World. New VersionRating: 5 out of 5 stars5/5 (51)
- The Three Waves of Volunteers & The New EarthFrom EverandThe Three Waves of Volunteers & The New EarthRating: 5 out of 5 stars5/5 (179)
- Meditations, On the Shortness of Life, The Enchiridion of Epictetus: The Ultimate Stoicism CollectionFrom EverandMeditations, On the Shortness of Life, The Enchiridion of Epictetus: The Ultimate Stoicism CollectionRating: 4.5 out of 5 stars4.5/5 (15)
- Summary: How to Know a Person: The Art of Seeing Others Deeply and Being Deeply Seen By David Brooks: Key Takeaways, Summary and AnalysisFrom EverandSummary: How to Know a Person: The Art of Seeing Others Deeply and Being Deeply Seen By David Brooks: Key Takeaways, Summary and AnalysisRating: 4 out of 5 stars4/5 (4)
- A Short History of Modern Philosophy: From Descartes to WittgensteinFrom EverandA Short History of Modern Philosophy: From Descartes to WittgensteinRating: 5 out of 5 stars5/5 (2)
- The Emperor's Handbook: A New Translation of The MeditationsFrom EverandThe Emperor's Handbook: A New Translation of The MeditationsRating: 4.5 out of 5 stars4.5/5 (9)
- Why Buddhism is True: The Science and Philosophy of Meditation and EnlightenmentFrom EverandWhy Buddhism is True: The Science and Philosophy of Meditation and EnlightenmentRating: 4.5 out of 5 stars4.5/5 (753)
- The Courage to Be Happy: Discover the Power of Positive Psychology and Choose Happiness Every DayFrom EverandThe Courage to Be Happy: Discover the Power of Positive Psychology and Choose Happiness Every DayRating: 4.5 out of 5 stars4.5/5 (215)
- How to Destroy America in Three Easy StepsFrom EverandHow to Destroy America in Three Easy StepsRating: 4.5 out of 5 stars4.5/5 (21)
- Summary of Man's Search for Meaning by Viktor E. FranklFrom EverandSummary of Man's Search for Meaning by Viktor E. FranklRating: 4.5 out of 5 stars4.5/5 (101)
- The Authoritarian Moment: How the Left Weaponized America's Institutions Against DissentFrom EverandThe Authoritarian Moment: How the Left Weaponized America's Institutions Against DissentRating: 4 out of 5 stars4/5 (17)
- It's Easier Than You Think: The Buddhist Way to HappinessFrom EverandIt's Easier Than You Think: The Buddhist Way to HappinessRating: 4 out of 5 stars4/5 (60)
- Moral Tribes: Emotion, Reason, and the Gap Between Us and ThemFrom EverandMoral Tribes: Emotion, Reason, and the Gap Between Us and ThemRating: 4.5 out of 5 stars4.5/5 (115)
- You Are Not Special: And Other EncouragementsFrom EverandYou Are Not Special: And Other EncouragementsRating: 4.5 out of 5 stars4.5/5 (6)
- Knocking on Heaven's Door: How Physics and Scientific Thinking Illuminate the Universe and the Modern WorldFrom EverandKnocking on Heaven's Door: How Physics and Scientific Thinking Illuminate the Universe and the Modern WorldRating: 3.5 out of 5 stars3.5/5 (64)