You might also like
- Shoe Dog: A Memoir by the Creator of NikeFrom EverandShoe Dog: A Memoir by the Creator of NikeRating: 4.5 out of 5 stars4.5/5 (537)
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeFrom EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeRating: 4 out of 5 stars4/5 (5794)
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceFrom EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceRating: 4 out of 5 stars4/5 (895)
- The Yellow House: A Memoir (2019 National Book Award Winner)From EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Rating: 4 out of 5 stars4/5 (98)
- Grit: The Power of Passion and PerseveranceFrom EverandGrit: The Power of Passion and PerseveranceRating: 4 out of 5 stars4/5 (588)
- The Little Book of Hygge: Danish Secrets to Happy LivingFrom EverandThe Little Book of Hygge: Danish Secrets to Happy LivingRating: 3.5 out of 5 stars3.5/5 (400)
- The Emperor of All Maladies: A Biography of CancerFrom EverandThe Emperor of All Maladies: A Biography of CancerRating: 4.5 out of 5 stars4.5/5 (271)
- Never Split the Difference: Negotiating As If Your Life Depended On ItFrom EverandNever Split the Difference: Negotiating As If Your Life Depended On ItRating: 4.5 out of 5 stars4.5/5 (838)
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyFrom EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyRating: 3.5 out of 5 stars3.5/5 (2259)
- On Fire: The (Burning) Case for a Green New DealFrom EverandOn Fire: The (Burning) Case for a Green New DealRating: 4 out of 5 stars4/5 (74)
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureFrom EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureRating: 4.5 out of 5 stars4.5/5 (474)
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryFrom EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryRating: 3.5 out of 5 stars3.5/5 (231)
- Team of Rivals: The Political Genius of Abraham LincolnFrom EverandTeam of Rivals: The Political Genius of Abraham LincolnRating: 4.5 out of 5 stars4.5/5 (234)
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaFrom EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaRating: 4.5 out of 5 stars4.5/5 (266)
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersFrom EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersRating: 4.5 out of 5 stars4.5/5 (345)
- The Unwinding: An Inner History of the New AmericaFrom EverandThe Unwinding: An Inner History of the New AmericaRating: 4 out of 5 stars4/5 (45)
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreFrom EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreRating: 4 out of 5 stars4/5 (1090)
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)From EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Rating: 4.5 out of 5 stars4.5/5 (121)
- Her Body and Other Parties: StoriesFrom EverandHer Body and Other Parties: StoriesRating: 4 out of 5 stars4/5 (821)
- PyrrophytaDocument34 pagesPyrrophytaMudasir Ahmad100% (1)
- Gene Cloning & Cloning Vectors: DR Ravi Kant Agrawal, MVSC, PHDDocument75 pagesGene Cloning & Cloning Vectors: DR Ravi Kant Agrawal, MVSC, PHDvivekNo ratings yet
- Lesson Plan Mitosis and MeiosisDocument9 pagesLesson Plan Mitosis and MeiosiscsamarinaNo ratings yet
- The Case of The Druid Dracula - PCR LabDocument12 pagesThe Case of The Druid Dracula - PCR LabTAUZIAH SUFINo ratings yet
- Pathology-Test-List For Christian 13022019Document73 pagesPathology-Test-List For Christian 13022019Mohanad EldarerNo ratings yet
- Highlights in Host Manipulation: ReviewDocument14 pagesHighlights in Host Manipulation: Reviewbing masturNo ratings yet
- Animal PhysiologyDocument104 pagesAnimal PhysiologyClara MaeNo ratings yet
- (Psychology of Emotions, Motivations and Actions) Leandro Cavalcanti (Ed.), Sofia Azevedo (Ed.) - Psychology of Stress - New Research-Nova Science Publishers (2013)Document214 pages(Psychology of Emotions, Motivations and Actions) Leandro Cavalcanti (Ed.), Sofia Azevedo (Ed.) - Psychology of Stress - New Research-Nova Science Publishers (2013)Jesus Jimenez100% (1)
- Edexcel Ial Biology Lab BookpdfDocument69 pagesEdexcel Ial Biology Lab Bookpdfbgtes12367% (6)
- Investigation of Toxic Effect of Moringa PDFDocument9 pagesInvestigation of Toxic Effect of Moringa PDFMiguel JuguilonNo ratings yet
- Hydrolysis of Nucleic Acids: Group 8Document25 pagesHydrolysis of Nucleic Acids: Group 8kiki parkNo ratings yet
- Bulletin 6060Document2 pagesBulletin 6060Muni SwamyNo ratings yet
- Bioprocess Considerations in Using Animal Cell CulturesDocument25 pagesBioprocess Considerations in Using Animal Cell CulturesDidem Kara100% (1)
- Cellular Respiration DefinitionDocument9 pagesCellular Respiration Definitionmaria genioNo ratings yet
- Journal of Genetic CounselingDocument23 pagesJournal of Genetic CounselingSaleemNo ratings yet
- MBoC5e - CHPT - 13 Intracellular Vesicular TrafficDocument111 pagesMBoC5e - CHPT - 13 Intracellular Vesicular TrafficAnnisaazharNo ratings yet
- Science BiologyCompleteDocument11 pagesScience BiologyCompleteJohanis Galaura EngcoNo ratings yet
- 108 PDFDocument7 pages108 PDFpuspa sariNo ratings yet
- ĐỀ ÔN TỈNH (N021) - HLK - TN - 2018 A. LISTENING (50 points) Urban Vs Suburban Living Advantages Disadvantages 1. - - - - - - - - - -Document7 pagesĐỀ ÔN TỈNH (N021) - HLK - TN - 2018 A. LISTENING (50 points) Urban Vs Suburban Living Advantages Disadvantages 1. - - - - - - - - - -Tran Anh ThaiNo ratings yet
- Supreme Court Monsanto Nuziveedu InvnTree Aug1stDocument9 pagesSupreme Court Monsanto Nuziveedu InvnTree Aug1stkira-auditoreNo ratings yet
- G 6 Scintbk 2Document275 pagesG 6 Scintbk 2Bullet Rubia100% (1)
- 18 Hour Chick EmbryoDocument7 pages18 Hour Chick Embryoaa628100% (2)
- Bab XIII Teknik Bioteknologi Untuk Penyelamatan Plasma Nutfah TanamanDocument37 pagesBab XIII Teknik Bioteknologi Untuk Penyelamatan Plasma Nutfah TanamanEben FernandoNo ratings yet
- Science COT 2 (4th Quarter)Document3 pagesScience COT 2 (4th Quarter)Rhazel Caballero100% (1)
- C1411-MagPure Particles NDocument1 pageC1411-MagPure Particles NZhang TimNo ratings yet
- mRNA Vaccines A New Era in VaccinologyDocument19 pagesmRNA Vaccines A New Era in Vaccinologyedysson1No ratings yet
- Definition, Aim, Objectives and Scope of Plant BreedingDocument137 pagesDefinition, Aim, Objectives and Scope of Plant BreedingYosef AndualemNo ratings yet
- Assignment No. 01: Human Digestive System and Respiratory SystemDocument3 pagesAssignment No. 01: Human Digestive System and Respiratory SystemZIA UR REHMANNo ratings yet
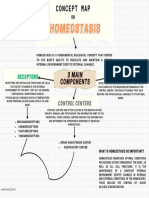
- Concept Map Group 2Document1 pageConcept Map Group 2mike jesterNo ratings yet
- Invertebrados Acelomados y PseudocelomadosDocument19 pagesInvertebrados Acelomados y PseudocelomadosPol PérezNo ratings yet