You might also like
- A-Level Chemistry Revision: Cheeky Revision ShortcutsFrom EverandA-Level Chemistry Revision: Cheeky Revision ShortcutsRating: 4 out of 5 stars4/5 (5)
- Chemical Physics Letters: Santanu Roy, Liem X. DangDocument5 pagesChemical Physics Letters: Santanu Roy, Liem X. Dangsyoran04No ratings yet
- Quantum Chemical Tests of Water-Water Potential For Inte Rac Tion Site Water ModelsDocument7 pagesQuantum Chemical Tests of Water-Water Potential For Inte Rac Tion Site Water ModelsRobost KostNo ratings yet
- Reevaluation of The Born Model Ion Hydration: 5588 Phys. Chem. 1985,89, 5588-5593Document6 pagesReevaluation of The Born Model Ion Hydration: 5588 Phys. Chem. 1985,89, 5588-5593Henry Kwame AgboviNo ratings yet
- Debye Huckel TheoryDocument19 pagesDebye Huckel TheoryJass Mattu100% (1)
- Electron Correlation: The Many-Body Problem at The Heart of ChemistryDocument14 pagesElectron Correlation: The Many-Body Problem at The Heart of ChemistryFranciscoNo ratings yet
- Liu-Prausnitz (1989) Thermodynamics of Concentrated Electrolyte SolutionsDocument25 pagesLiu-Prausnitz (1989) Thermodynamics of Concentrated Electrolyte SolutionsPolanqNo ratings yet
- 29 - CHAPTER 3 Intermolecular Forces and Potential Enegy SurfacesDocument9 pages29 - CHAPTER 3 Intermolecular Forces and Potential Enegy SurfacesMohit Kamboj100% (2)
- Propensity of Heavier Halides For The Water/vapor Interface Revisited Using The Amoeba Force FieldDocument14 pagesPropensity of Heavier Halides For The Water/vapor Interface Revisited Using The Amoeba Force FieldDominik JeníčekNo ratings yet
- Quantum Coherent Water PDFDocument9 pagesQuantum Coherent Water PDFVera Gardasevic MitrovicNo ratings yet
- Science 2010 Larsen 65 9Document6 pagesScience 2010 Larsen 65 9MycoLogist4LifeNo ratings yet
- Teemu Salmi Et Al - Calculation of The O-H Stretching Vibrational Overtone Spectrum of The Water DimerDocument8 pagesTeemu Salmi Et Al - Calculation of The O-H Stretching Vibrational Overtone Spectrum of The Water Dimer4534567No ratings yet
- Unifying Solution and Surface Electrochemistry: Limitations and Opportunities in Surface ElectrocatalysisDocument7 pagesUnifying Solution and Surface Electrochemistry: Limitations and Opportunities in Surface ElectrocatalysisMiguel Angel SandovalNo ratings yet
- OCR Chemistry A Level Y2 GlossaryDocument14 pagesOCR Chemistry A Level Y2 GlossaryifratsubhaNo ratings yet
- Water Chemistry BybtyuDocument8 pagesWater Chemistry BybtyutulsiNo ratings yet
- PRL 2013 Bocquet Nanofluidic Osmotic Diodes Theory and Molecular Dynamics SimulationsDocument5 pagesPRL 2013 Bocquet Nanofluidic Osmotic Diodes Theory and Molecular Dynamics SimulationsChirodeep BakliNo ratings yet
- Physical ChenistryDocument146 pagesPhysical ChenistrychemasimNo ratings yet
- SKB Journal 4.Document28 pagesSKB Journal 4.anasstem2025No ratings yet
- Functionalized Nanomaterial Based Electrochemical Sensors Principles Fabrication Methods and Applications Chaudhery Mustansar Hussain 2 Full ChapterDocument52 pagesFunctionalized Nanomaterial Based Electrochemical Sensors Principles Fabrication Methods and Applications Chaudhery Mustansar Hussain 2 Full Chapterdustin.erickson563100% (12)
- Article Valorisation Eaux GrisesDocument12 pagesArticle Valorisation Eaux GrisesGérardNo ratings yet
- An Equation of State For Electrolyte Solutions Covering Wide Ranges of Temperature, Pressure, and CompositionDocument16 pagesAn Equation of State For Electrolyte Solutions Covering Wide Ranges of Temperature, Pressure, and CompositionSaleh SedighiNo ratings yet
- The ANANKE Relative Energy Gradient (REG) Method To Automate IQA Analysis Over Configurational ChangeDocument13 pagesThe ANANKE Relative Energy Gradient (REG) Method To Automate IQA Analysis Over Configurational ChangeRikardo Pino RiosNo ratings yet
- 14 Chapter 2Document65 pages14 Chapter 2chemsac2No ratings yet
- Ab Initio Molecular Dynamics Simulation of Hydrogen DiffusionDocument4 pagesAb Initio Molecular Dynamics Simulation of Hydrogen DiffusionzhuhanjiNo ratings yet
- Molecular Physics: Please Scroll Down For ArticleDocument8 pagesMolecular Physics: Please Scroll Down For Article4534567No ratings yet
- IECR98 WaterDocument10 pagesIECR98 WaterpeNo ratings yet
- A Case of Negative Apparent Activation Energy Due To Pore Diffusion EffectsDocument3 pagesA Case of Negative Apparent Activation Energy Due To Pore Diffusion EffectsBamrung SungnoenNo ratings yet
- Signature Properties of Water: Their Molecular Electronic OriginsDocument6 pagesSignature Properties of Water: Their Molecular Electronic Origins丁竑維No ratings yet
- Numerical Investigations On Ethanol Electrolysis For Production of Pure Hydrogen From Renewable SourcesDocument6 pagesNumerical Investigations On Ethanol Electrolysis For Production of Pure Hydrogen From Renewable SourcesSanchez JorgeNo ratings yet
- Dynamics of Reactions in Polar Solvents. Semiclassical Trajectory Studies of Electron-Transfer and Proton-Transfer ReactionsDocument7 pagesDynamics of Reactions in Polar Solvents. Semiclassical Trajectory Studies of Electron-Transfer and Proton-Transfer ReactionsMoh SyaifudinNo ratings yet
- C 3 RedoxDocument28 pagesC 3 RedoxRina P ShresthaNo ratings yet
- Ebook Functionalized Nanomaterial Based Electrochemical Sensors Principles Fabrication Methods and Applications 2 Full Chapter PDFDocument68 pagesEbook Functionalized Nanomaterial Based Electrochemical Sensors Principles Fabrication Methods and Applications 2 Full Chapter PDFbertha.senior550100% (24)
- The Nature of The Chemonuclear Transition Hidetsugu IkegamiDocument55 pagesThe Nature of The Chemonuclear Transition Hidetsugu IkegamigarofasNo ratings yet
- Bonding III.1-7Document7 pagesBonding III.1-7Kartik DuttaNo ratings yet
- 1903 10054Document20 pages1903 10054Naveen PrakashNo ratings yet
- CV ExperimentDocument8 pagesCV ExperimentesatpehlivanNo ratings yet
- Water Molecule StructureDocument9 pagesWater Molecule StructureKezial Angel100% (1)
- Mathematical Models of Distribution of Water Molecules Regarding Energies of Hydrogen BondsDocument25 pagesMathematical Models of Distribution of Water Molecules Regarding Energies of Hydrogen BondsjrNo ratings yet
- TMP BE95Document7 pagesTMP BE95FrontiersNo ratings yet
- 4.07 Ions in Aqueous Solution: (5 Points)Document9 pages4.07 Ions in Aqueous Solution: (5 Points)NadunKodikaraNo ratings yet
- C11 20 PDFDocument11 pagesC11 20 PDFOscar A. LuévanoNo ratings yet
- J.electrochem - Soc. 1991 Springer 2334 42Document9 pagesJ.electrochem - Soc. 1991 Springer 2334 42DEVA NAIKNo ratings yet
- Course Code 6457: Assignment No. 1Document10 pagesCourse Code 6457: Assignment No. 1Tahirullah KhanNo ratings yet
- 34 - О - Моделирование и симуляция пузырькового течения H 2 -H 2 O через пакет из трех ячеек в предпилотном реакторе электрокоагуляции фильтр-прессаDocument12 pages34 - О - Моделирование и симуляция пузырькового течения H 2 -H 2 O через пакет из трех ячеек в предпилотном реакторе электрокоагуляции фильтр-прессаТатьянаNo ratings yet
- On The Nature and Signatures of The Solvated Electron in WaterDocument13 pagesOn The Nature and Signatures of The Solvated Electron in WaterMycoLogist4LifeNo ratings yet
- Learning Goals Unit 3 and 4Document6 pagesLearning Goals Unit 3 and 4Sana SyedNo ratings yet
- 1407 3162 PDFDocument97 pages1407 3162 PDFAnonymous 80p9OVNo ratings yet
- We Are Intechopen, The World'S Leading Publisher of Open Access Books Built by Scientists, For ScientistsDocument18 pagesWe Are Intechopen, The World'S Leading Publisher of Open Access Books Built by Scientists, For ScientistsJean Carlos Jimenez HuillcaNo ratings yet
- Project 2Document9 pagesProject 2Nikhil KumarNo ratings yet
- Virial Equation of StateDocument9 pagesVirial Equation of StateSaba ArifNo ratings yet
- STC 111 PDFDocument34 pagesSTC 111 PDFogbonna ebuka innocentNo ratings yet
- We Are Intechopen, The World'S Leading Publisher of Open Access Books Built by Scientists, For ScientistsDocument18 pagesWe Are Intechopen, The World'S Leading Publisher of Open Access Books Built by Scientists, For ScientistslucasNo ratings yet
- Some Thermodynamic Properties of The Hydrated Electron: Joshua Jortner RichardDocument5 pagesSome Thermodynamic Properties of The Hydrated Electron: Joshua Jortner RichardMycoLogist4LifeNo ratings yet
- Alex Kaivarainen - New Hierarchic Theory of Water and Its Role in Biosystems: The Quantum Psi ProblemDocument54 pagesAlex Kaivarainen - New Hierarchic Theory of Water and Its Role in Biosystems: The Quantum Psi ProblemJoaokaaNo ratings yet
- Elements From The SeaDocument11 pagesElements From The SeaLaurenNo ratings yet
- Chapter 4Document18 pagesChapter 4Umesh ChandraNo ratings yet
- Hydrogen AtomDocument8 pagesHydrogen AtomElyasse B.No ratings yet
- Quantum ChemistryDocument13 pagesQuantum ChemistryAnsh PooniaNo ratings yet
- TorDocument11 pagesTorమత్సా చంద్ర శేఖర్No ratings yet
- Shape-Chick-2d-Shape-Activity-Sheets Ver 1Document3 pagesShape-Chick-2d-Shape-Activity-Sheets Ver 1api-279675519No ratings yet
- 10 1021@je301082yDocument10 pages10 1021@je301082yమత్సా చంద్ర శేఖర్No ratings yet
- Coherence (Physics) - WikipediaDocument10 pagesCoherence (Physics) - Wikipediaమత్సా చంద్ర శేఖర్No ratings yet
- 09 Chapter 2Document35 pages09 Chapter 2మత్సా చంద్ర శేఖర్No ratings yet
- 1401 6814 PDFDocument22 pages1401 6814 PDFమత్సా చంద్ర శేఖర్No ratings yet
- Physics 11 Part 1Document255 pagesPhysics 11 Part 1kalaikalai360No ratings yet
- A 1Document6 pagesA 1మత్సా చంద్ర శేఖర్No ratings yet
- 10 1016@j Chemphys 2016 05 028Document11 pages10 1016@j Chemphys 2016 05 028మత్సా చంద్ర శేఖర్No ratings yet
- Doing Physics With Matlab Mathematical Routines: Solving Quadratic EquationsDocument2 pagesDoing Physics With Matlab Mathematical Routines: Solving Quadratic Equationsమత్సా చంద్ర శేఖర్No ratings yet
- Reciprocal Lattice FCC BCC SCDocument19 pagesReciprocal Lattice FCC BCC SCnikhiltembhare_60052No ratings yet
- Adsorption of He Gas On The Agn Nanoclusters A Molecular Dynamic Study 2014 Fluid Phase EquilibriaDocument5 pagesAdsorption of He Gas On The Agn Nanoclusters A Molecular Dynamic Study 2014 Fluid Phase Equilibriaమత్సా చంద్ర శేఖర్No ratings yet
- Articles of Chandra SekharDocument1 pageArticles of Chandra Sekharమత్సా చంద్ర శేఖర్No ratings yet
- SSP Hilke PDFDocument34 pagesSSP Hilke PDFTùng Danh NguyễnNo ratings yet
- Adsorption Isotherms and Surface Geometry 2001 Fluid Phase EquilibriaDocument9 pagesAdsorption Isotherms and Surface Geometry 2001 Fluid Phase Equilibriaమత్సా చంద్ర శేఖర్No ratings yet
- OptDocument253 pagesOpttictac4136063No ratings yet
- Eur J Inorg Chem 2010 1717Document12 pagesEur J Inorg Chem 2010 1717మత్సా చంద్ర శేఖర్No ratings yet
- JfigsdgjdthjDocument7 pagesJfigsdgjdthjDanilo PedrelliNo ratings yet
- Kim 2013Document7 pagesKim 2013మత్సా చంద్ర శేఖర్No ratings yet
- 10 1016@j Chemphys 2016 05 028Document11 pages10 1016@j Chemphys 2016 05 028మత్సా చంద్ర శేఖర్No ratings yet
- Els 2Document9 pagesEls 2మత్సా చంద్ర శేఖర్No ratings yet
- Els 3pdDocument5 pagesEls 3pdమత్సా చంద్ర శేఖర్No ratings yet
- Fulltext PDFDocument24 pagesFulltext PDFమత్సా చంద్ర శేఖర్100% (1)
- Lecture 2Document13 pagesLecture 2Neelam KapoorNo ratings yet
- Lecture On Vector CalculusDocument2 pagesLecture On Vector Calculusspaul4uNo ratings yet
- Math 123Document47 pagesMath 123ShailendraPatelNo ratings yet
- 1 Vector CalculusDocument16 pages1 Vector Calculusమత్సా చంద్ర శేఖర్No ratings yet
- SLV 2Document22 pagesSLV 2Anirban DasNo ratings yet
- How To Best Explain Divergence and Curl - QuoraDocument12 pagesHow To Best Explain Divergence and Curl - Quoraమత్సా చంద్ర శేఖర్No ratings yet
- Gas Laws K L Kapoor McGraw Hill JEE AdvancedDocument44 pagesGas Laws K L Kapoor McGraw Hill JEE Advancedshubhang2392No ratings yet
- Richard Becker (Auth.) - Theory of Heat-Springer Berlin Heidelberg (1967) PDFDocument393 pagesRichard Becker (Auth.) - Theory of Heat-Springer Berlin Heidelberg (1967) PDFJulian SierraNo ratings yet
- The Maxwell Relations: CHEM 331 Physical Chemistry Fall 2017Document11 pagesThe Maxwell Relations: CHEM 331 Physical Chemistry Fall 2017AhmedovicMichaelNo ratings yet
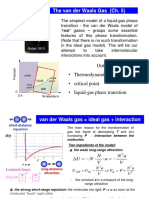
- Lecture 16. The Van Der Waals Gas (Ch. 5)Document25 pagesLecture 16. The Van Der Waals Gas (Ch. 5)sandesh gaikwadNo ratings yet
- Geofluids - 2009 - BAKKER - Package FLUIDS Part 3 Correlations Between Equations of State Thermodynamics and FluidDocument12 pagesGeofluids - 2009 - BAKKER - Package FLUIDS Part 3 Correlations Between Equations of State Thermodynamics and FluidMustapha MaaroufNo ratings yet
- IPS May 2004Document15 pagesIPS May 2004Obumse Chukwuebuka MichaelNo ratings yet
- 4211 Sheet 3Document2 pages4211 Sheet 3Roy VeseyNo ratings yet
- Thermodynamics FullDocument105 pagesThermodynamics FullSravan SingireddyNo ratings yet
- Expansion VirialDocument52 pagesExpansion VirialAlejandro CardenasNo ratings yet
- Homework F PHY3513: RT PVDocument2 pagesHomework F PHY3513: RT PVBob BelcherNo ratings yet
- Accumulator, Hydraulic - EquationsDocument7 pagesAccumulator, Hydraulic - Equationsrsproserv0% (1)
- Van Der Waals Equation Devation From Ideal Gas Laws: Notes Prepared by Prof DR Hikmat S. Al-SalimDocument14 pagesVan Der Waals Equation Devation From Ideal Gas Laws: Notes Prepared by Prof DR Hikmat S. Al-SalimSàtz ÑÖÑïtNo ratings yet
- LT1 G7Document22 pagesLT1 G7Chelsea MartinezNo ratings yet
- 18 Tugas Sifat Sifat Gas CH 1Document19 pages18 Tugas Sifat Sifat Gas CH 1Fadhillah AnsyariNo ratings yet
- Soave-Redlich-Kwong Adiabatic Equation For Gas-Loaded AccumulatorDocument7 pagesSoave-Redlich-Kwong Adiabatic Equation For Gas-Loaded AccumulatorlucasNo ratings yet
- ps1 QuestionsDocument2 pagesps1 QuestionsdjambulazizNo ratings yet
- Chapter 5 Gases PDFDocument49 pagesChapter 5 Gases PDFAbou WalidNo ratings yet
- TDA 301T - 2015-10 - Thermodynamic Properties Real SubstancesDocument146 pagesTDA 301T - 2015-10 - Thermodynamic Properties Real SubstancesMduduzi Magiva Mahlangu100% (1)
- Intro To MathcadDocument13 pagesIntro To Mathcadrmelo2010No ratings yet
- Diffuse Interface Models in Fluid Mechanics: Didier Jamet CEA-GrenobleDocument35 pagesDiffuse Interface Models in Fluid Mechanics: Didier Jamet CEA-GrenobleSubramanya SadasivaNo ratings yet
- Gaseous State (Real Gas) ExerciseDocument13 pagesGaseous State (Real Gas) ExercisemikcNo ratings yet
- States of MatterDocument17 pagesStates of MatterTEJAS CHHABRANo ratings yet
- Theoretical BiophysicsDocument211 pagesTheoretical Biophysicsanil ariNo ratings yet
- Fiziks: Chapter - 2Document13 pagesFiziks: Chapter - 2SURAJ PRATAP SINGHNo ratings yet
- Assignment 1Document2 pagesAssignment 1Prince Mensah100% (2)
- Wa0001.Document31 pagesWa0001.Stefani KavangoNo ratings yet
- Topic 1 - Introduction To Critical Phenomena and Phase Transitions - Course - Advanced Statistical Mechanics - 1er Sem - 2020Document8 pagesTopic 1 - Introduction To Critical Phenomena and Phase Transitions - Course - Advanced Statistical Mechanics - 1er Sem - 2020karen romanNo ratings yet
- Equations of State PDFDocument3 pagesEquations of State PDFRoozbeh PNo ratings yet
- Ideal GasDocument17 pagesIdeal GasPoonamNo ratings yet
- AP® Chemistry Crash Course, For the 2020 Exam, Book + Online: Get a Higher Score in Less TimeFrom EverandAP® Chemistry Crash Course, For the 2020 Exam, Book + Online: Get a Higher Score in Less TimeRating: 5 out of 5 stars5/5 (1)
- Periodic Tales: A Cultural History of the Elements, from Arsenic to ZincFrom EverandPeriodic Tales: A Cultural History of the Elements, from Arsenic to ZincRating: 3.5 out of 5 stars3.5/5 (137)
- Is That a Fact?: Frauds, Quacks, and the Real Science of Everyday LifeFrom EverandIs That a Fact?: Frauds, Quacks, and the Real Science of Everyday LifeRating: 5 out of 5 stars5/5 (4)
- The Nature of Drugs Vol. 1: History, Pharmacology, and Social ImpactFrom EverandThe Nature of Drugs Vol. 1: History, Pharmacology, and Social ImpactRating: 5 out of 5 stars5/5 (5)
- Handbook of Formulating Dermal Applications: A Definitive Practical GuideFrom EverandHandbook of Formulating Dermal Applications: A Definitive Practical GuideNo ratings yet
- Fundamentals of Chemistry: A Modern IntroductionFrom EverandFundamentals of Chemistry: A Modern IntroductionRating: 5 out of 5 stars5/5 (1)
- AP Chemistry Flashcards, Fourth Edition: Up-to-Date Review and PracticeFrom EverandAP Chemistry Flashcards, Fourth Edition: Up-to-Date Review and PracticeNo ratings yet
- Formulating, Packaging, and Marketing of Natural Cosmetic ProductsFrom EverandFormulating, Packaging, and Marketing of Natural Cosmetic ProductsNo ratings yet
- Organic Chemistry for Schools: Advanced Level and Senior High SchoolFrom EverandOrganic Chemistry for Schools: Advanced Level and Senior High SchoolNo ratings yet
- Chemistry for Breakfast: The Amazing Science of Everyday LifeFrom EverandChemistry for Breakfast: The Amazing Science of Everyday LifeRating: 4.5 out of 5 stars4.5/5 (90)
- Monkeys, Myths, and Molecules: Separating Fact from Fiction, and the Science of Everyday LifeFrom EverandMonkeys, Myths, and Molecules: Separating Fact from Fiction, and the Science of Everyday LifeRating: 4 out of 5 stars4/5 (1)
- Tribology: Friction and Wear of Engineering MaterialsFrom EverandTribology: Friction and Wear of Engineering MaterialsRating: 5 out of 5 stars5/5 (1)
- The Periodic Table: A Very Short IntroductionFrom EverandThe Periodic Table: A Very Short IntroductionRating: 4.5 out of 5 stars4.5/5 (3)
- Ingredients: A Visual Exploration of 75 Additives & 25 Food ProductsFrom EverandIngredients: A Visual Exploration of 75 Additives & 25 Food ProductsRating: 4 out of 5 stars4/5 (1)
- Taste: Surprising Stories and Science About Why Food Tastes GoodFrom EverandTaste: Surprising Stories and Science About Why Food Tastes GoodRating: 3 out of 5 stars3/5 (20)
- The Elements We Live By: How Iron Helps Us Breathe, Potassium Lets Us See, and Other Surprising Superpowers of the Periodic TableFrom EverandThe Elements We Live By: How Iron Helps Us Breathe, Potassium Lets Us See, and Other Surprising Superpowers of the Periodic TableRating: 3.5 out of 5 stars3.5/5 (22)
- The Disappearing Spoon: And Other True Tales of Madness, Love, and the History of the World from the Periodic Table of the ElementsFrom EverandThe Disappearing Spoon: And Other True Tales of Madness, Love, and the History of the World from the Periodic Table of the ElementsRating: 4 out of 5 stars4/5 (146)
- Bioplastics: A Home Inventors HandbookFrom EverandBioplastics: A Home Inventors HandbookRating: 4 out of 5 stars4/5 (2)
- Guidelines for Defining Process Safety Competency RequirementsFrom EverandGuidelines for Defining Process Safety Competency RequirementsRating: 3 out of 5 stars3/5 (1)
- The Nature of Drugs Vol. 1: History, Pharmacology, and Social ImpactFrom EverandThe Nature of Drugs Vol. 1: History, Pharmacology, and Social ImpactRating: 5 out of 5 stars5/5 (1)
- Chemistry for Breakfast: The Amazing Science of Everyday LifeFrom EverandChemistry for Breakfast: The Amazing Science of Everyday LifeRating: 4.5 out of 5 stars4.5/5 (14)
- The Regenerative Grower's Guide to Garden Amendments: Using Locally Sourced Materials to Make Mineral and Biological Extracts and FermentsFrom EverandThe Regenerative Grower's Guide to Garden Amendments: Using Locally Sourced Materials to Make Mineral and Biological Extracts and FermentsRating: 5 out of 5 stars5/5 (3)