You might also like
- Computational Methods for Process SimulationFrom EverandComputational Methods for Process SimulationRating: 3 out of 5 stars3/5 (1)
- Don't Gamble With Physical Properties For SimulationsDocument12 pagesDon't Gamble With Physical Properties For Simulationslaiping_lum100% (1)
- Chemical Engineering Calculations Lecture Notes 1: Introduction To Basic Concepts and CalculationsDocument7 pagesChemical Engineering Calculations Lecture Notes 1: Introduction To Basic Concepts and CalculationsTaanzNo ratings yet
- Paper 28Document8 pagesPaper 28Hassan Funsho AkandeNo ratings yet
- System Modelling & Simulation of Energy SystemsDocument35 pagesSystem Modelling & Simulation of Energy SystemsH.J.PrabhuNo ratings yet
- Modeling Notes CompleteDocument144 pagesModeling Notes CompleteHarsha100% (1)
- Thermodynamic ModelsDocument24 pagesThermodynamic ModelsSampathkumar AttuluriNo ratings yet
- Distillation Column ModellingDocument1 pageDistillation Column ModellingChem.EnggNo ratings yet
- Bequette 1 Y2Document25 pagesBequette 1 Y2Kami JaraNo ratings yet
- Marlin Ch03 Process ControlsDocument47 pagesMarlin Ch03 Process ControlsRagavaaRagavaa100% (1)
- Chapter 8. Process ModelingDocument21 pagesChapter 8. Process Modelingdebjit123No ratings yet
- Oredo2Swlpl) Dwlrqri&Khplfdo (Qjlqhhulqj 3Odqwve/Phdqvri (Yroxwlrqdu/$OjrulwkpvDocument13 pagesOredo2Swlpl) Dwlrqri&Khplfdo (Qjlqhhulqj 3Odqwve/Phdqvri (Yroxwlrqdu/$OjrulwkpvMadhavanIceNo ratings yet
- Aspen Hysys SimulationDocument5 pagesAspen Hysys SimulationEngr Mohammad FarhanNo ratings yet
- Theoretical Lectures 1&2 (PM&S) - 7 Semester: Introduction To Computer-Aided Process Design and SimulationDocument8 pagesTheoretical Lectures 1&2 (PM&S) - 7 Semester: Introduction To Computer-Aided Process Design and Simulationyasa osmanNo ratings yet
- Control Degrees of FreedomDocument11 pagesControl Degrees of FreedomAlastair WongNo ratings yet
- Process Corrosion Simulation PaperDocument19 pagesProcess Corrosion Simulation PaperMohammad Fouladi100% (1)
- Catalysts: Process Simulation For The Design and Scale Up of Heterogeneous Catalytic Process: Kinetic Modelling IssuesDocument33 pagesCatalysts: Process Simulation For The Design and Scale Up of Heterogeneous Catalytic Process: Kinetic Modelling Issuesjesus de jesusNo ratings yet
- Rate Based Distillation Williums PaperDocument4 pagesRate Based Distillation Williums Paperzorro21072107No ratings yet
- Mathematical Modeling and Numerical Analysis MScDocument163 pagesMathematical Modeling and Numerical Analysis MScZaidoon MohsinNo ratings yet
- Mechatronic System Design Optimisation WorkshopDocument9 pagesMechatronic System Design Optimisation WorkshopMradul YadavNo ratings yet
- FM Assignment 1Document4 pagesFM Assignment 1Narender SinghNo ratings yet
- Property Package Selection Tree - Processdesign Mccormick (Good)Document12 pagesProperty Package Selection Tree - Processdesign Mccormick (Good)daraj darajNo ratings yet
- Pages9 53Document45 pagesPages9 53Hana HamidNo ratings yet
- Selecting Thermodynamic Models For Process Simulation of Organic VLE and LLE SystemsDocument5 pagesSelecting Thermodynamic Models For Process Simulation of Organic VLE and LLE SystemsoscarmaumarNo ratings yet
- The Process Simulation Revolution: Thermophysical Property Needs and ConcernsDocument4 pagesThe Process Simulation Revolution: Thermophysical Property Needs and ConcernsbjsatolaNo ratings yet
- Distillation Dynamics and Control Workbook 2006 PDFDocument18 pagesDistillation Dynamics and Control Workbook 2006 PDFEr Mayur PatilNo ratings yet
- AIAA-2000-1437 Validation of Structural Dynamics Models at Los Alamos National LaboratoryDocument15 pagesAIAA-2000-1437 Validation of Structural Dynamics Models at Los Alamos National LaboratoryRomoex R RockNo ratings yet
- Future Foods: Coding Caviar To Avoid Species ExtinctionDocument4 pagesFuture Foods: Coding Caviar To Avoid Species ExtinctionLadsNo ratings yet
- Applied Thermodynamics For Process ModelingDocument7 pagesApplied Thermodynamics For Process ModelingdhavalmpNo ratings yet
- Overview of Mechanistic Modelling TechniquesDocument6 pagesOverview of Mechanistic Modelling TechniquesGXGGXGNo ratings yet
- Generalized Reservoir Modeling WorkflowDocument5 pagesGeneralized Reservoir Modeling Workflowروان كامل زمام حسن التميميNo ratings yet
- Simulation of Process Lines in Microsoft Excel - Nitric Acid ProductionDocument16 pagesSimulation of Process Lines in Microsoft Excel - Nitric Acid ProductionmanojNo ratings yet
- Integrate Process Simulation and Process SynthesisDocument6 pagesIntegrate Process Simulation and Process Synthesisjanota24No ratings yet
- Process Simulation For Distillation DesignDocument34 pagesProcess Simulation For Distillation DesignLenin PrabhuNo ratings yet
- Designing a Workable SystemDocument8 pagesDesigning a Workable SystemMichael ChikwenduNo ratings yet
- 1.2.2 Thermodynamics and HYSYS - 5 PDFDocument26 pages1.2.2 Thermodynamics and HYSYS - 5 PDFWelisson SilvaNo ratings yet
- 0708 0369Document27 pages0708 0369iledinamo1No ratings yet
- HSC Chemistry 8Document12 pagesHSC Chemistry 8Lubomira Macheva100% (1)
- Chemical Process Simulation: by Dr. Ch. KavithaDocument16 pagesChemical Process Simulation: by Dr. Ch. KavithaVenkata GiriNo ratings yet
- New StylesDocument4 pagesNew StylesKarimPrinceAddoNo ratings yet
- Thermodynamic Models & Physical Properties: Property Method SelectionDocument24 pagesThermodynamic Models & Physical Properties: Property Method SelectiondoufethiNo ratings yet
- NTNU C3-MR Process Dynamic Modeling and Control SimulationDocument52 pagesNTNU C3-MR Process Dynamic Modeling and Control SimulationSrihari Kodimela75% (4)
- HYSYS Methanol ProductionDocument77 pagesHYSYS Methanol ProductionGaukharAlzhanova100% (1)
- Mathematical Models and Algorithms for Power System Optimization: Modeling Technology for Practical Engineering ProblemsFrom EverandMathematical Models and Algorithms for Power System Optimization: Modeling Technology for Practical Engineering ProblemsNo ratings yet
- Laboratory Statistics: Methods in Chemistry and Health SciencesFrom EverandLaboratory Statistics: Methods in Chemistry and Health SciencesNo ratings yet
- Modeling and Simulation of Thermal Power Plants with ThermoSysPro: A Theoretical Introduction and a Practical GuideFrom EverandModeling and Simulation of Thermal Power Plants with ThermoSysPro: A Theoretical Introduction and a Practical GuideNo ratings yet
- Dimensional Analysis: Practical Guides in Chemical EngineeringFrom EverandDimensional Analysis: Practical Guides in Chemical EngineeringNo ratings yet
- Measurement and Control Basics, 4th EditionFrom EverandMeasurement and Control Basics, 4th EditionRating: 4 out of 5 stars4/5 (11)
- Weighted Residual Methods: Principles, Modifications and ApplicationsFrom EverandWeighted Residual Methods: Principles, Modifications and ApplicationsNo ratings yet
- Computer-Controlled Systems: Theory and Design, Third EditionFrom EverandComputer-Controlled Systems: Theory and Design, Third EditionRating: 3 out of 5 stars3/5 (4)
- Chemical Engineering Process SimulationFrom EverandChemical Engineering Process SimulationRating: 4 out of 5 stars4/5 (13)
- Digital Control Engineering: Analysis and DesignFrom EverandDigital Control Engineering: Analysis and DesignRating: 3 out of 5 stars3/5 (1)
- FoglerDocument12 pagesFoglerDzaky FajratamaNo ratings yet
- Bazilian2013 2Document10 pagesBazilian2013 2Dzaky FajratamaNo ratings yet
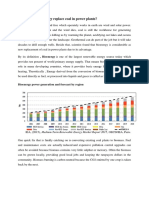
- Is It Possible Bioenergy Replace Coal in Power Plants?Document3 pagesIs It Possible Bioenergy Replace Coal in Power Plants?Dzaky FajratamaNo ratings yet
- Transport PhenomenaDocument13 pagesTransport PhenomenaDzaky FajratamaNo ratings yet
- Vapor Pressure of A Liquid SolutionDocument40 pagesVapor Pressure of A Liquid Solutionintania660% (1)
- Shellsol 2046 Ar: Technical DatasheetDocument3 pagesShellsol 2046 Ar: Technical DatasheetJavier Godoy MirandaNo ratings yet
- UNIT-2 Abstactions and RunoffDocument17 pagesUNIT-2 Abstactions and RunoffKushaal RajNo ratings yet
- Datasheet Isoparaffinsshellsoltdeurope PDFDocument3 pagesDatasheet Isoparaffinsshellsoltdeurope PDFmeNo ratings yet
- Thermodynamic Review: Chemical Thermodynamics Exam 2 GuideDocument9 pagesThermodynamic Review: Chemical Thermodynamics Exam 2 GuideFaizan AhmedNo ratings yet
- Aplicacion de Sowtfare para I.Q.Document34 pagesAplicacion de Sowtfare para I.Q.Gabriel MenchuNo ratings yet
- Secado y Deshidratación de AlimentosDocument37 pagesSecado y Deshidratación de Alimentoseduardo tafurNo ratings yet
- TD MODULE 5Document30 pagesTD MODULE 5mujeebNo ratings yet
- Unit4 1 Fluid StaticsDocument30 pagesUnit4 1 Fluid StaticsANSHU RAJNo ratings yet
- Refrigerant Recover/Recycling Equipment: 1998 Standard ForDocument21 pagesRefrigerant Recover/Recycling Equipment: 1998 Standard ForMazen MohamedNo ratings yet
- Handout Chapter 17Document30 pagesHandout Chapter 17Sam H. SalehNo ratings yet
- Lev JKKP PDFDocument53 pagesLev JKKP PDFMohamad AsrulNo ratings yet
- l4 The Clausius-UpadiDocument8 pagesl4 The Clausius-UpadiMarcos Sánchez MartínezNo ratings yet
- Specific Heat of Liquids and Solids: Standard Test Method ForDocument6 pagesSpecific Heat of Liquids and Solids: Standard Test Method ForJean-Patrice DeliaNo ratings yet
- D130 PDFDocument6 pagesD130 PDFJuan Diego ArizabalNo ratings yet
- Fluid Properties: Density, Specific Volume, Specific Weight, Specific Gravity, and PressureDocument1 pageFluid Properties: Density, Specific Volume, Specific Weight, Specific Gravity, and PressuresolidwormNo ratings yet
- Astm d2029 Dew PTDocument6 pagesAstm d2029 Dew PTvlcmstne04No ratings yet
- A Fundamental Equation For Calculation of The Thermodynamic Properties of EthanolDocument15 pagesA Fundamental Equation For Calculation of The Thermodynamic Properties of EthanolFernando AraujoNo ratings yet
- Thermodynamic PropertiesDocument44 pagesThermodynamic PropertiesVellia AzoraNo ratings yet
- DIG Wash and Block Buffer Set 1 PC: Safety Data SheetDocument21 pagesDIG Wash and Block Buffer Set 1 PC: Safety Data SheetGEINER ANDRES OSSA GALVIS0% (1)
- Chemistry Investigatory Project AnshDocument19 pagesChemistry Investigatory Project Anshlalithadithya85No ratings yet
- Ch. 11: Liquids and Intermolecular Forces: - GasesDocument22 pagesCh. 11: Liquids and Intermolecular Forces: - GasesJozel Bryan Mestiola TerrìbleNo ratings yet
- 2 Phase Relief Calculations Spreadsheet: Basis General DataDocument1 page2 Phase Relief Calculations Spreadsheet: Basis General DataAffian WidjanarkoNo ratings yet
- MSA Gas Detection HandbookDocument146 pagesMSA Gas Detection HandbookVid YullNo ratings yet
- Thermodynamics Enthalpy Entropy Mollier and Steam TablesDocument44 pagesThermodynamics Enthalpy Entropy Mollier and Steam TablesashisNo ratings yet
- Irrigation Design Pipe HydraulicsDocument47 pagesIrrigation Design Pipe Hydraulicshadush gebreNo ratings yet
- Liquid Solutions PDFDocument50 pagesLiquid Solutions PDFAniruddha KawadeNo ratings yet
- PNG 520 - Course On Phase RelationsDocument172 pagesPNG 520 - Course On Phase RelationsDenstar Ricardo SilalahiNo ratings yet
- Thermodynamics of Physical and Chemical Vapour Deposition: 2.1 Ideal GasesDocument31 pagesThermodynamics of Physical and Chemical Vapour Deposition: 2.1 Ideal GasesHarry Fernando SembiringNo ratings yet