You might also like
- Assignment Clo1 EnergyDocument3 pagesAssignment Clo1 EnergyaNo ratings yet
- Liquid Liquid Extraction in A Packed BedDocument26 pagesLiquid Liquid Extraction in A Packed BedAr SaidNo ratings yet
- Eni Completion Procedures ManualDocument269 pagesEni Completion Procedures ManualMahmoud Ahmed Ali Abdelrazik100% (1)
- Lab Petchem (Exp 4)Document13 pagesLab Petchem (Exp 4)Hafiz ImranNo ratings yet
- DistillationDocument124 pagesDistillationasharab70100% (1)
- Distillation 3Document36 pagesDistillation 3Renu SekaranNo ratings yet
- Supercritical Fluid Extraction of Ethanol From Aqueous SolutionsDocument11 pagesSupercritical Fluid Extraction of Ethanol From Aqueous SolutionsPedro LisboaNo ratings yet
- Chee3004 Unit Operations Mccabe-Thiele Method & Multicomponent DistillationDocument14 pagesChee3004 Unit Operations Mccabe-Thiele Method & Multicomponent Distillationking4lifeNo ratings yet
- Fractionation and Absorption ExplainedDocument26 pagesFractionation and Absorption ExplainedmohamedNo ratings yet
- Chapter 4 - Efficiency of ColumnsDocument38 pagesChapter 4 - Efficiency of ColumnsAnonymous vRU4VlNo ratings yet
- 474 - CHM 703Document25 pages474 - CHM 703permata100% (1)
- Distillation 2Document20 pagesDistillation 2arslanadeelNo ratings yet
- Distillation Notes 2011Document40 pagesDistillation Notes 2011samuelgranthamNo ratings yet
- Pressure-Swing Reactive Distillation Process For Transesterification of Methyl Acetate With IsopropanolDocument3 pagesPressure-Swing Reactive Distillation Process For Transesterification of Methyl Acetate With IsopropanolMahesh ChantarkarNo ratings yet
- Effect of Two-Pass Trays On Distillation EfficienciesDocument7 pagesEffect of Two-Pass Trays On Distillation EfficienciesCheng ZehanNo ratings yet
- Multicomponent Distillation OptimizationDocument9 pagesMulticomponent Distillation OptimizationSriparthan SriramanNo ratings yet
- RI Vs Composition Methanol-Water MixtureDocument12 pagesRI Vs Composition Methanol-Water MixtureAnonymous VeJYFSMWLINo ratings yet
- Distillation Experiment LehighDocument10 pagesDistillation Experiment Lehighcgjp120391No ratings yet
- 7-Liquid Liquid Extraction - COMPLETEDocument39 pages7-Liquid Liquid Extraction - COMPLETERickyWisaksonoNo ratings yet
- Distillation TheoryDocument40 pagesDistillation TheoryIrvin HernandezNo ratings yet
- Chapter - 1Document9 pagesChapter - 1Sahil PatilNo ratings yet
- Introduction To Separation Process EngineeringDocument12 pagesIntroduction To Separation Process EngineeringAna K. CalderónNo ratings yet
- Spray Drying Process ExplainedDocument3 pagesSpray Drying Process ExplainedParadise CatNo ratings yet
- Fluidized Bed Experiment AnalysisDocument11 pagesFluidized Bed Experiment AnalysisHaiqal AzizNo ratings yet
- Packed Distillation Column ExperimentDocument20 pagesPacked Distillation Column ExperimentChan Chun ChenNo ratings yet
- Batch Distillation Experiment AnalysisDocument5 pagesBatch Distillation Experiment AnalysisNurul Atikah JapryNo ratings yet
- Lab Manual MainDocument59 pagesLab Manual MainYusof Kaizer100% (1)
- Distillation Principles ExplainedDocument71 pagesDistillation Principles ExplainedMuhammad Qaisar KhanNo ratings yet
- Azeotropes VLE DataDocument17 pagesAzeotropes VLE Datamehul10941No ratings yet
- Chemical Reactor Types: PFRs and CSTRsDocument2 pagesChemical Reactor Types: PFRs and CSTRsWancianSiaNo ratings yet
- Rate Based Vs Equilibrium ModelDocument12 pagesRate Based Vs Equilibrium Modelzorro21072107No ratings yet
- The General Structure of A Chemical ProcessDocument16 pagesThe General Structure of A Chemical Processagrocel_bhv5591No ratings yet
- Supercritical CO2 Extraction of Helichrysum ItalicumDocument8 pagesSupercritical CO2 Extraction of Helichrysum ItalicumTaipe SollerNo ratings yet
- CHE545 MASS TRANSFER ASSIGNMENTDocument3 pagesCHE545 MASS TRANSFER ASSIGNMENTsoapNo ratings yet
- Fluidized bed reactor design and fabricationDocument11 pagesFluidized bed reactor design and fabricationHarshaNo ratings yet
- Armfield Ht31 Tubular Heat Exchanger in The Education KeywordsDocument3 pagesArmfield Ht31 Tubular Heat Exchanger in The Education KeywordsCHERUYIOT IAN100% (1)
- Mass Transfer Unit Operations in Food ProcessingDocument1 pageMass Transfer Unit Operations in Food ProcessingGenet0% (1)
- 5.2. Classification of FuelsDocument16 pages5.2. Classification of FuelsadiNo ratings yet
- Separation Processes - I (CHE F244) Total Marks - 15 Due Date & Time: 01/07/2020, 5:00 PM AssignmentDocument4 pagesSeparation Processes - I (CHE F244) Total Marks - 15 Due Date & Time: 01/07/2020, 5:00 PM AssignmentElliot AldersonNo ratings yet
- Bioreactor Scale-UpDocument12 pagesBioreactor Scale-Uphsiegler2No ratings yet
- Modelling The Extraction of Essential Oils With Compressed Carbon DioxideDocument14 pagesModelling The Extraction of Essential Oils With Compressed Carbon DioxideYusiana DewiNo ratings yet
- Absorption Lecture Note - DR Akinsiku PDFDocument7 pagesAbsorption Lecture Note - DR Akinsiku PDFGlory UsoroNo ratings yet
- Mass Transfer Coefficient ExplainedDocument37 pagesMass Transfer Coefficient ExplainednivedhithaNo ratings yet
- Crystallization (Latest)Document31 pagesCrystallization (Latest)Lin Xian XingNo ratings yet
- Extraction of sesame seed oil using supercritical CO2 and mathematical modelingDocument7 pagesExtraction of sesame seed oil using supercritical CO2 and mathematical modelingJonatas LopesNo ratings yet
- Separation Processss Lecture NotesDocument17 pagesSeparation Processss Lecture NoteskeatyNo ratings yet
- 42-Single Effect Evaporator-21-May-2021Material I 21-May-2021 Single Effect EvaporatorDocument8 pages42-Single Effect Evaporator-21-May-2021Material I 21-May-2021 Single Effect EvaporatorAbhishek KarpeNo ratings yet
- Chemical Engineering Equations and ConceptsDocument12 pagesChemical Engineering Equations and ConceptsAbhishek PadmasaleNo ratings yet
- Carbondioxide ScrubberDocument8 pagesCarbondioxide ScrubberSameer ChalkeNo ratings yet
- 05 Web Mansoori AsphaltenesDocument18 pages05 Web Mansoori AsphaltenesjardelbrunoNo ratings yet
- Vapor Liquid EquilibriumDocument39 pagesVapor Liquid EquilibriumyeenNo ratings yet
- Liquid LiquidDocument8 pagesLiquid LiquidAnonymous b9fcR5No ratings yet
- PP-309 Mass Transfer: Course Facilitator: Nadia Khan Lecture of Week 1 & 2Document77 pagesPP-309 Mass Transfer: Course Facilitator: Nadia Khan Lecture of Week 1 & 2ashas waseem100% (1)
- Exp-9 - Liquid Liquid Extraction in A Packed ColumnDocument5 pagesExp-9 - Liquid Liquid Extraction in A Packed ColumnSiddharth MohapatraNo ratings yet
- Rauolt's LawDocument11 pagesRauolt's LawSagar JunejaNo ratings yet
- Applications of AdsorptionDocument6 pagesApplications of AdsorptionmrshashmiNo ratings yet
- Coulson and Richardson’s Chemical Engineering: Volume 2B: Separation ProcessesFrom EverandCoulson and Richardson’s Chemical Engineering: Volume 2B: Separation ProcessesAjay Kumar RayNo ratings yet
- Insights into Chemical Engineering: Selected Papers of P.V. DanckwertsFrom EverandInsights into Chemical Engineering: Selected Papers of P.V. DanckwertsNo ratings yet
- Distillation IDocument57 pagesDistillation IOsan ThorpeNo ratings yet
- Chapter Two Distillation ProcessDocument80 pagesChapter Two Distillation ProcessMatewos SadaNo ratings yet
- CHE333 Simultaneous Heat & Mass Transfer Operations Lecture 4: DistillationDocument59 pagesCHE333 Simultaneous Heat & Mass Transfer Operations Lecture 4: DistillationB MasoomNo ratings yet
- Text Book and Notes Grading: Homework, Test, Midterm and FinalDocument127 pagesText Book and Notes Grading: Homework, Test, Midterm and FinalChristian NwekeNo ratings yet
- DISTILLATION UNIT 1 28.2.22 - WatermarkDocument26 pagesDISTILLATION UNIT 1 28.2.22 - WatermarkHardik ChauhanNo ratings yet
- Transport Phenomena (Transfer Processes)Document38 pagesTransport Phenomena (Transfer Processes)esiri aluyaNo ratings yet
- Pre Treatment Methods in The Production of BiodieselDocument3 pagesPre Treatment Methods in The Production of BiodieselemmanuelNo ratings yet
- TO Philosophy, Logic and Human ExistenceDocument9 pagesTO Philosophy, Logic and Human ExistenceemmanuelNo ratings yet
- 1Document28 pages1emmanuelNo ratings yet
- TO Philosophy, Logic and Human ExistenceDocument9 pagesTO Philosophy, Logic and Human ExistenceemmanuelNo ratings yet
- ValueDocument1 pageValueemmanuelNo ratings yet
- CHE413 Note2Document40 pagesCHE413 Note2emmanuelNo ratings yet
- Name: Omonjade Emmanuel MATRIC NO: 15CF02658 Department: Chemical EngineeringDocument2 pagesName: Omonjade Emmanuel MATRIC NO: 15CF02658 Department: Chemical EngineeringemmanuelNo ratings yet
- Che 416 Fundamentals For Part-A 2018-19 SessionDocument12 pagesChe 416 Fundamentals For Part-A 2018-19 SessionemmanuelNo ratings yet
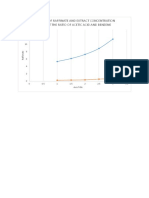
- A Graph of Raffinate and Extract Concentration Against The Ratio of Acetic Acid and BenzeneDocument1 pageA Graph of Raffinate and Extract Concentration Against The Ratio of Acetic Acid and BenzeneemmanuelNo ratings yet
- Graph of Conductivity Against Time After Impulse Change For Reactor 2Document9 pagesGraph of Conductivity Against Time After Impulse Change For Reactor 2emmanuelNo ratings yet
- Name: Omonjade Emmanuel MATRIC NO: 15CF02658 Department: Chemical EngineeringDocument2 pagesName: Omonjade Emmanuel MATRIC NO: 15CF02658 Department: Chemical EngineeringemmanuelNo ratings yet
- Discussion of Result and CoclusionDocument2 pagesDiscussion of Result and CoclusionemmanuelNo ratings yet
- CHEMICAL REACTION ENGINEERING COURSE OUTLINEDocument81 pagesCHEMICAL REACTION ENGINEERING COURSE OUTLINEemmanuel100% (1)
- Che 326 Lecture NotesDocument155 pagesChe 326 Lecture Noteswinifred ekpoNo ratings yet
- Che 326 Lecture NotesDocument155 pagesChe 326 Lecture Noteswinifred ekpoNo ratings yet
- Assignment I - Module 3 (CHE320)Document2 pagesAssignment I - Module 3 (CHE320)NelsonNo ratings yet
- Che 321 Power Pt.Document42 pagesChe 321 Power Pt.emmanuelNo ratings yet
- Fire Technology and Arson InvestigationDocument18 pagesFire Technology and Arson InvestigationKiven M. GeonzonNo ratings yet
- RAC Lecture 13Document25 pagesRAC Lecture 13Mohammed SiddiqueNo ratings yet
- Harris Regulator ManualDocument12 pagesHarris Regulator ManualButton DavidsonNo ratings yet
- Seminar Nasional Kimia KALTIM (Prof. Dr. Hadi Nur)Document58 pagesSeminar Nasional Kimia KALTIM (Prof. Dr. Hadi Nur)VeronikaSantiMarbunNo ratings yet
- Compresores Hermticos Tecumseh Lunite Hermetique 5 17062015Document1 pageCompresores Hermticos Tecumseh Lunite Hermetique 5 17062015rafael velandi velandiNo ratings yet
- Fischer TropschDocument64 pagesFischer TropschHoracio RodriguezNo ratings yet
- Heterogeneous Catalysis Processes and ApplicationsDocument16 pagesHeterogeneous Catalysis Processes and ApplicationshamoodahNo ratings yet
- Detailed Design For Yas Mina Zayed Pipeline Project: Class-I (Category-I) Documents Control RegisterDocument186 pagesDetailed Design For Yas Mina Zayed Pipeline Project: Class-I (Category-I) Documents Control RegisterSuresh SelvamNo ratings yet
- MKT.503 - Elliott Turbomachinery For Refineries PDFDocument8 pagesMKT.503 - Elliott Turbomachinery For Refineries PDFMilan TrengovskiNo ratings yet
- Corrosion Specialist or Corrosion EngineerDocument2 pagesCorrosion Specialist or Corrosion Engineerapi-77851650No ratings yet
- MIG Welding - Gas Metal Arc Welding (GMAW)Document5 pagesMIG Welding - Gas Metal Arc Welding (GMAW)Mohamed AtefNo ratings yet
- BP Toledo v6 LoDocument20 pagesBP Toledo v6 LoMarcos ArraezNo ratings yet
- Chapter 9 and 10Document22 pagesChapter 9 and 10Paolo GochingcoNo ratings yet
- K Series JIC Tube Fittings PDFDocument40 pagesK Series JIC Tube Fittings PDFtree4dhanNo ratings yet
- 12Document16 pages12Anonymous kBjvdERRQNo ratings yet
- Oil-Free Compression of Syngas White Paper 6.24.20Document4 pagesOil-Free Compression of Syngas White Paper 6.24.20JoséNo ratings yet
- Underground Gas StorageDocument8 pagesUnderground Gas StorageAhmed KhairyNo ratings yet
- Ap Unit7 Worksheet AnswersDocument5 pagesAp Unit7 Worksheet Answersburcak gecNo ratings yet
- LAS-Gen.Chem2_MELC_20-22_Q3-Week-8Document11 pagesLAS-Gen.Chem2_MELC_20-22_Q3-Week-8JV Subang PatindolNo ratings yet
- Engineering Encyclopedia: Exxon Chemical and Mechanical Cleaning Manual 6) Precommission Cleaning and PassivationDocument14 pagesEngineering Encyclopedia: Exxon Chemical and Mechanical Cleaning Manual 6) Precommission Cleaning and Passivationcvg ertdNo ratings yet
- Introduction To Petroleum EngineeringDocument52 pagesIntroduction To Petroleum EngineeringJane Eaton100% (2)
- Water SplittingDocument7 pagesWater SplittingRommel BalceNo ratings yet
- Line Identification Line P&Id Size Pipe No. Line No. (In.) Spec Service (PD-) FromDocument56 pagesLine Identification Line P&Id Size Pipe No. Line No. (In.) Spec Service (PD-) Fromsamer8saifNo ratings yet
- IGCSE Chemistry: Chemical Reactions - NotesDocument13 pagesIGCSE Chemistry: Chemical Reactions - NotesNiharikaNo ratings yet
- Gas Compression 101Document41 pagesGas Compression 101Mormor OmertaNo ratings yet
- Distillation Exp.Document5 pagesDistillation Exp.Ibrahim DewaliNo ratings yet
- ACRE2a Non Elementary Reaction Kinetics RevDocument53 pagesACRE2a Non Elementary Reaction Kinetics RevRathish RagooNo ratings yet