You might also like
- Answers to Selected Problems in Multivariable Calculus with Linear Algebra and SeriesFrom EverandAnswers to Selected Problems in Multivariable Calculus with Linear Algebra and SeriesRating: 1.5 out of 5 stars1.5/5 (2)
- Lecture 4: PID of A Spring Mass Damper SystemDocument6 pagesLecture 4: PID of A Spring Mass Damper SystemAditya AdNo ratings yet
- Field-angle dependence of thermal transport in Kitaev-Γ modelDocument4 pagesField-angle dependence of thermal transport in Kitaev-Γ modelのすをNo ratings yet
- Assignment #1: 1 HgjyygbykvrfDocument1 pageAssignment #1: 1 HgjyygbykvrfJuan Sebastian ArangoNo ratings yet
- Nuggets: Evaluating Rate Laws: 3. Integrated Rate Laws, 4. Half-Lives, 5. Rate Versus ConcentrationDocument12 pagesNuggets: Evaluating Rate Laws: 3. Integrated Rate Laws, 4. Half-Lives, 5. Rate Versus ConcentrationRosario QFNo ratings yet
- Quality Planning Tools1Document40 pagesQuality Planning Tools1agungNo ratings yet
- 6 Pdfsam 2111.00854Document1 page6 Pdfsam 2111.00854knjkjnkkj 99No ratings yet
- Degree Electronic Engineeringe First Semester EXAMINATIONS 2020/2021Document3 pagesDegree Electronic Engineeringe First Semester EXAMINATIONS 2020/2021Peter JumreNo ratings yet
- MTE119 - Solutions Hw2Document9 pagesMTE119 - Solutions Hw2fabioNo ratings yet
- One-Way Slab Design ProcedureDocument2 pagesOne-Way Slab Design Procedurethescubatater0% (1)
- Feedback Control - On Off & PIDDocument20 pagesFeedback Control - On Off & PIDRizaldi MaulanaNo ratings yet
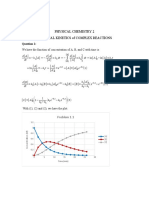
- Chem Kinetic (Complex)Document5 pagesChem Kinetic (Complex)Trung VõNo ratings yet
- Seismic Blending and Deblending in 3DDocument5 pagesSeismic Blending and Deblending in 3DFatma MAGRAOUINo ratings yet
- Analogous SystemsDocument2 pagesAnalogous SystemsDhruv KanthaliyaNo ratings yet
- ECE2202 Final Exam Fall2017Document29 pagesECE2202 Final Exam Fall2017अजय ढकालNo ratings yet
- Objective Physics For NEET Vol 1-Pages-4Document8 pagesObjective Physics For NEET Vol 1-Pages-4StockPlusIndiaNo ratings yet
- Chemical Kinetics: CHEM. 204Document17 pagesChemical Kinetics: CHEM. 204myriamNo ratings yet
- Solution Manual For Fluid Mechanics For Engineers by ChinDocument32 pagesSolution Manual For Fluid Mechanics For Engineers by China1246048300% (1)
- ESE-2018 Mains Test Series: Mechanical Engineering Test No: 14Document40 pagesESE-2018 Mains Test Series: Mechanical Engineering Test No: 14ravi kumar kumawatNo ratings yet
- b14 SP Lect3Document22 pagesb14 SP Lect3Pubg MobilNo ratings yet
- Week-8 Assignment8 DBMSDocument10 pagesWeek-8 Assignment8 DBMSRoshanNo ratings yet
- TEE QP B11 MAT3002 Applied Linear AlgebraDocument2 pagesTEE QP B11 MAT3002 Applied Linear Algebra4013Bodkhe VaishnoviNo ratings yet
- Week 8 Assignment and SolutionsDocument7 pagesWeek 8 Assignment and SolutionsG. Dancer GhNo ratings yet
- Kinetics LectureDocument10 pagesKinetics LectureJose GamezNo ratings yet
- Ps 3Document3 pagesPs 3baoke renNo ratings yet
- Data Structures KCS 301 1 2021Document2 pagesData Structures KCS 301 1 2021Hariom JadonNo ratings yet
- Assessment BriefDocument8 pagesAssessment BriefShahrukh KhanNo ratings yet
- Homework 3Document5 pagesHomework 3Prakhar AgrawalNo ratings yet
- ESE-2018 Mains Test Series: Mechanical Engineering Test No: 14Document40 pagesESE-2018 Mains Test Series: Mechanical Engineering Test No: 14VivekMishraNo ratings yet
- Second Mid-Term Examination (23%) Vii Sem, 2018 - 19Document2 pagesSecond Mid-Term Examination (23%) Vii Sem, 2018 - 19Kartik ModiNo ratings yet
- Mid Sem Exam 2021-22Document2 pagesMid Sem Exam 2021-22N Rabindra PatraNo ratings yet
- Isometric: Table of MaterialsDocument3 pagesIsometric: Table of MaterialsilijarskNo ratings yet
- Series de PuseuixDocument13 pagesSeries de PuseuixGabrielBenícioNo ratings yet
- Quiz 1Document1 pageQuiz 1Kishor KunalNo ratings yet
- Validation Case: Flow Past A Cylinder: November 19, 2018Document3 pagesValidation Case: Flow Past A Cylinder: November 19, 2018hieuleeNo ratings yet
- Ans & Sol - JEE (Main) - 2022 - Phase-2 - 26-07-2022 - Evening - (Physics)Document6 pagesAns & Sol - JEE (Main) - 2022 - Phase-2 - 26-07-2022 - Evening - (Physics)Ashish SrivastavaNo ratings yet
- Semester 1 Examinations 2018/2019: Programme (S)Document5 pagesSemester 1 Examinations 2018/2019: Programme (S)fuckoffmanNo ratings yet
- Moment Distribution For BeamsDocument8 pagesMoment Distribution For BeamsMichaella Bianca De GuzmanNo ratings yet
- Ps 2Document3 pagesPs 2baoke renNo ratings yet
- Finite Element AnalysisDocument9 pagesFinite Element AnalysisKING AHK GAMINGNo ratings yet
- Problema 3Document8 pagesProblema 3andersson rondinel cartolinNo ratings yet
- Tutorial - 5 and Solution Feb 9 2017 FinalDocument2 pagesTutorial - 5 and Solution Feb 9 2017 FinalRounak Majumdar100% (1)
- Exam121022 SolDocument6 pagesExam121022 Solchiedzad madondoNo ratings yet
- Trefethen BauDocument29 pagesTrefethen BauHari Haran100% (1)
- MATH39032Document6 pagesMATH39032Luke MillerNo ratings yet
- Column-Design EthiopianDocument8 pagesColumn-Design EthiopianMesfin100% (2)
- The Capacity Spectrum Method As A Tool For Seismic DesignDocument8 pagesThe Capacity Spectrum Method As A Tool For Seismic DesignNIE100% (2)
- Column DesignAAUDocument5 pagesColumn DesignAAUEngineeri TadiyosNo ratings yet
- Solo6 50Document19 pagesSolo6 50AndyNo ratings yet
- Mte360 W08 MT1Document2 pagesMte360 W08 MT1dholedaysNo ratings yet
- l6 PDFDocument11 pagesl6 PDFdanielNo ratings yet
- Column Design Column-C1: M M M MDocument3 pagesColumn Design Column-C1: M M M MMustefa Mohammed AdemNo ratings yet
- 2.JEE Main 2019 Jan 9 Forenoon Session Answer Key SolutionDocument26 pages2.JEE Main 2019 Jan 9 Forenoon Session Answer Key Solutiongp1in007No ratings yet
- Cbse X.maths Term I Practice PaperDocument5 pagesCbse X.maths Term I Practice PaperSach BalwaniNo ratings yet
- Convection Sample ProblemDocument1 pageConvection Sample ProblemGenerale, Rey marck C.No ratings yet
- Topic 3Document10 pagesTopic 3Nima DorjiNo ratings yet
- Chemical KinematicsDocument3 pagesChemical KinematicsLyra GurimbaoNo ratings yet
- Lab 6 - AnswersDocument9 pagesLab 6 - AnswersEdivaldo BartolomeuNo ratings yet
- ME 380 Chapter 2 HW Solution: Review QuestionsDocument5 pagesME 380 Chapter 2 HW Solution: Review QuestionsVisakan ParameswaranNo ratings yet
- Nano MaterialsDocument38 pagesNano Materialsaedi0611100% (3)
- Table 6.2. Acidity of Some Hydrocarbons: Entry Hydrocarbon Cs (CHA) Cs (THF) K (DMSO)Document1 pageTable 6.2. Acidity of Some Hydrocarbons: Entry Hydrocarbon Cs (CHA) Cs (THF) K (DMSO)panda biruNo ratings yet
- Engineering Physics Questions and AnswersDocument95 pagesEngineering Physics Questions and AnswersSheambom Nelson100% (1)
- Manfred Held - Liners For Shaped ChargesDocument7 pagesManfred Held - Liners For Shaped ChargesGhoree23456No ratings yet
- Chem Exp. 6 Chemical EquilibriumDocument15 pagesChem Exp. 6 Chemical EquilibriumRachel MaguireNo ratings yet
- Cantilever Beam Experiment B58SBDocument9 pagesCantilever Beam Experiment B58SBSunNo ratings yet
- dmos특성 결과표Document49 pagesdmos특성 결과표고경진No ratings yet
- Hazardous Waste ManagementDocument20 pagesHazardous Waste ManagementYuki SalemNo ratings yet
- Stas ReviewDocument35 pagesStas Reviewchristine manaloNo ratings yet
- Green ChemistryDocument12 pagesGreen ChemistrymidhunNo ratings yet
- Goulds e SH Stainless Steel Pumps Technical Brochure BeSH R2Document60 pagesGoulds e SH Stainless Steel Pumps Technical Brochure BeSH R2Daniel Ortiz GonzálezNo ratings yet
- Answer 589Document3 pagesAnswer 589Sasa LiliNo ratings yet
- Pressure Vessel Design by Biruk Solomon BadeDocument76 pagesPressure Vessel Design by Biruk Solomon BadebrookNo ratings yet
- Pressure Control Group Surface Treatment Specification Per NORSOKDocument33 pagesPressure Control Group Surface Treatment Specification Per NORSOKevenNo ratings yet
- Fram AW32 Hydr Fluid SDS 7-17-15Document9 pagesFram AW32 Hydr Fluid SDS 7-17-15AbhimanyaNo ratings yet
- Ketene Production and Utilization Experimental StudyDocument4 pagesKetene Production and Utilization Experimental Studyspedhome1No ratings yet
- The Bungee Jump: Potential Energy at Work: Ais ChallengeDocument20 pagesThe Bungee Jump: Potential Energy at Work: Ais ChallengeCIVILNo ratings yet
- PX01X XXX XXX Axxx enDocument12 pagesPX01X XXX XXX Axxx enPatrick BrilhanteNo ratings yet
- Year 5 Science Test PaperDocument13 pagesYear 5 Science Test Paperemelliaghalli75% (52)
- As Chemistry Unit 2 NotesDocument26 pagesAs Chemistry Unit 2 NotesArchitNo ratings yet
- Regenerator Reflux Pump - 2L1x2-10ARV 1Document4 pagesRegenerator Reflux Pump - 2L1x2-10ARV 1Efril dilen franciscoNo ratings yet
- 83120-On The Dynamics of The Friction CoefficientDocument7 pages83120-On The Dynamics of The Friction CoefficientVitor JardimNo ratings yet
- CompassDocument139 pagesCompassErick Olano100% (1)
- Degumming, Refining, Bleaching, and Deodorization TheoryDocument2 pagesDegumming, Refining, Bleaching, and Deodorization TheoryNityantiniNo ratings yet
- The Optical Properties of Magnesium Oxide ContainiDocument6 pagesThe Optical Properties of Magnesium Oxide ContainiAbdulbar kelilNo ratings yet
- Practical WS 6, 7 and 8 - AnswersDocument20 pagesPractical WS 6, 7 and 8 - AnswersrrrNo ratings yet
- Free Space and Ray Tracing ModelsDocument11 pagesFree Space and Ray Tracing ModelsDatta RajendraNo ratings yet
- Em SyllabusDocument6 pagesEm SyllabusSuresh BalamNo ratings yet
- Chem Topic 1 QuestionsDocument27 pagesChem Topic 1 QuestionsOscarHigson-SpenceNo ratings yet
- BETCK105 Eset 1Document2 pagesBETCK105 Eset 1Tejas krishnakanthNo ratings yet
- Process Plant Equipment: Operation, Control, and ReliabilityFrom EverandProcess Plant Equipment: Operation, Control, and ReliabilityRating: 5 out of 5 stars5/5 (1)
- Lees' Process Safety Essentials: Hazard Identification, Assessment and ControlFrom EverandLees' Process Safety Essentials: Hazard Identification, Assessment and ControlRating: 4 out of 5 stars4/5 (4)
- Process Steam Systems: A Practical Guide for Operators, Maintainers, and DesignersFrom EverandProcess Steam Systems: A Practical Guide for Operators, Maintainers, and DesignersNo ratings yet
- Guidelines for Chemical Process Quantitative Risk AnalysisFrom EverandGuidelines for Chemical Process Quantitative Risk AnalysisRating: 5 out of 5 stars5/5 (1)
- Sodium Bicarbonate: Nature's Unique First Aid RemedyFrom EverandSodium Bicarbonate: Nature's Unique First Aid RemedyRating: 5 out of 5 stars5/5 (21)
- The Perfumed Pages of History: A Textbook on Fragrance CreationFrom EverandThe Perfumed Pages of History: A Textbook on Fragrance CreationRating: 4 out of 5 stars4/5 (1)
- An Applied Guide to Water and Effluent Treatment Plant DesignFrom EverandAn Applied Guide to Water and Effluent Treatment Plant DesignRating: 5 out of 5 stars5/5 (4)
- Water-Based Paint Formulations, Vol. 3From EverandWater-Based Paint Formulations, Vol. 3Rating: 4.5 out of 5 stars4.5/5 (6)
- Functional Safety from Scratch: A Practical Guide to Process Industry ApplicationsFrom EverandFunctional Safety from Scratch: A Practical Guide to Process Industry ApplicationsNo ratings yet
- Well Control for Completions and InterventionsFrom EverandWell Control for Completions and InterventionsRating: 4 out of 5 stars4/5 (10)
- Pulp and Paper Industry: Emerging Waste Water Treatment TechnologiesFrom EverandPulp and Paper Industry: Emerging Waste Water Treatment TechnologiesRating: 5 out of 5 stars5/5 (1)
- Distillation Design and Control Using Aspen SimulationFrom EverandDistillation Design and Control Using Aspen SimulationRating: 5 out of 5 stars5/5 (2)
- Fun Facts about Hydrogen : Chemistry for Kids The Element Series | Children's Chemistry BooksFrom EverandFun Facts about Hydrogen : Chemistry for Kids The Element Series | Children's Chemistry BooksNo ratings yet
- Chemical Engineering Design: Principles, Practice and Economics of Plant and Process DesignFrom EverandChemical Engineering Design: Principles, Practice and Economics of Plant and Process DesignRating: 4 out of 5 stars4/5 (16)
- A New Approach to HAZOP of Complex Chemical ProcessesFrom EverandA New Approach to HAZOP of Complex Chemical ProcessesNo ratings yet
- The Periodic Table of Elements - Halogens, Noble Gases and Lanthanides and Actinides | Children's Chemistry BookFrom EverandThe Periodic Table of Elements - Halogens, Noble Gases and Lanthanides and Actinides | Children's Chemistry BookNo ratings yet
- Handbook of Cosmetic Science: An Introduction to Principles and ApplicationsFrom EverandHandbook of Cosmetic Science: An Introduction to Principles and ApplicationsH. W. HibbottRating: 4 out of 5 stars4/5 (6)
- Nuclear Energy in the 21st Century: World Nuclear University PressFrom EverandNuclear Energy in the 21st Century: World Nuclear University PressRating: 4.5 out of 5 stars4.5/5 (3)
- Bioinspired Materials Science and EngineeringFrom EverandBioinspired Materials Science and EngineeringGuang YangNo ratings yet
- The Stress Analysis of Pressure Vessels and Pressure Vessel Components: International Series of Monographs in Mechanical EngineeringFrom EverandThe Stress Analysis of Pressure Vessels and Pressure Vessel Components: International Series of Monographs in Mechanical EngineeringS. S. GillRating: 3.5 out of 5 stars3.5/5 (3)
- Coupled CFD-DEM Modeling: Formulation, Implementation and Application to Multiphase FlowsFrom EverandCoupled CFD-DEM Modeling: Formulation, Implementation and Application to Multiphase FlowsNo ratings yet
- Fundamentals of Risk Management for Process Industry EngineersFrom EverandFundamentals of Risk Management for Process Industry EngineersNo ratings yet
- Cathodic Protection: Industrial Solutions for Protecting Against CorrosionFrom EverandCathodic Protection: Industrial Solutions for Protecting Against CorrosionNo ratings yet