You might also like
- Atorvastatin Loaded Nanostructured Lipid Carriers NLCs Strategy To Overcome Oral Delivery DrawbacksDocument11 pagesAtorvastatin Loaded Nanostructured Lipid Carriers NLCs Strategy To Overcome Oral Delivery Drawbacksanh23No ratings yet
- DyslipidemiaDocument87 pagesDyslipidemiaCarlos Arturo Paternina Cuello100% (1)
- 1 s2.0 S0260877407000726 Main PDFDocument8 pages1 s2.0 S0260877407000726 Main PDFanh23No ratings yet
- Chapter 8Document14 pagesChapter 8Nghi NguyenNo ratings yet
- 2017 GINA Report, Global Strategy For Asthma Management and Prevention PDFDocument159 pages2017 GINA Report, Global Strategy For Asthma Management and Prevention PDFNalemi JT100% (1)
- (Vnguitar - Net) - Dong Thoai (Tong Hua - Guang Liang) Solo (Tone C - Ban Sua Lai) PDFDocument4 pages(Vnguitar - Net) - Dong Thoai (Tong Hua - Guang Liang) Solo (Tone C - Ban Sua Lai) PDFanh23No ratings yet
- Good Distribution Practices Trs 957 Annex 5Document30 pagesGood Distribution Practices Trs 957 Annex 5felicityNo ratings yet
- (Vnguitar - Net) - Dong Thoai (Tong Hua - Guang Liang) Solo (Tone C - Ban Sua Lai) PDFDocument4 pages(Vnguitar - Net) - Dong Thoai (Tong Hua - Guang Liang) Solo (Tone C - Ban Sua Lai) PDFanh23No ratings yet
- Dyslipidemia ATP4 GUIDLINESDocument9 pagesDyslipidemia ATP4 GUIDLINESSandy GunawanNo ratings yet
- Good Distribution Practices Trs 957 Annex 5Document30 pagesGood Distribution Practices Trs 957 Annex 5felicityNo ratings yet
- Waiver of In Vivo Studies for IR Solid Oral Dosage FormsDocument19 pagesWaiver of In Vivo Studies for IR Solid Oral Dosage Formsanh23No ratings yet
- Grand-Piano 01 I e A4Document7 pagesGrand-Piano 01 I e A4Géimar Marín ToroNo ratings yet
- Doctor Job Application LetterDocument2 pagesDoctor Job Application Letteranh23No ratings yet
- Shoe Dog: A Memoir by the Creator of NikeFrom EverandShoe Dog: A Memoir by the Creator of NikeRating: 4.5 out of 5 stars4.5/5 (537)
- Grit: The Power of Passion and PerseveranceFrom EverandGrit: The Power of Passion and PerseveranceRating: 4 out of 5 stars4/5 (587)
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceFrom EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceRating: 4 out of 5 stars4/5 (890)
- The Yellow House: A Memoir (2019 National Book Award Winner)From EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Rating: 4 out of 5 stars4/5 (98)
- The Little Book of Hygge: Danish Secrets to Happy LivingFrom EverandThe Little Book of Hygge: Danish Secrets to Happy LivingRating: 3.5 out of 5 stars3.5/5 (399)
- On Fire: The (Burning) Case for a Green New DealFrom EverandOn Fire: The (Burning) Case for a Green New DealRating: 4 out of 5 stars4/5 (73)
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeFrom EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeRating: 4 out of 5 stars4/5 (5794)
- Never Split the Difference: Negotiating As If Your Life Depended On ItFrom EverandNever Split the Difference: Negotiating As If Your Life Depended On ItRating: 4.5 out of 5 stars4.5/5 (838)
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureFrom EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureRating: 4.5 out of 5 stars4.5/5 (474)
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryFrom EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryRating: 3.5 out of 5 stars3.5/5 (231)
- The Emperor of All Maladies: A Biography of CancerFrom EverandThe Emperor of All Maladies: A Biography of CancerRating: 4.5 out of 5 stars4.5/5 (271)
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreFrom EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreRating: 4 out of 5 stars4/5 (1090)
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyFrom EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyRating: 3.5 out of 5 stars3.5/5 (2219)
- Team of Rivals: The Political Genius of Abraham LincolnFrom EverandTeam of Rivals: The Political Genius of Abraham LincolnRating: 4.5 out of 5 stars4.5/5 (234)
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersFrom EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersRating: 4.5 out of 5 stars4.5/5 (344)
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaFrom EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaRating: 4.5 out of 5 stars4.5/5 (265)
- The Unwinding: An Inner History of the New AmericaFrom EverandThe Unwinding: An Inner History of the New AmericaRating: 4 out of 5 stars4/5 (45)
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)From EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Rating: 4.5 out of 5 stars4.5/5 (119)
- Her Body and Other Parties: StoriesFrom EverandHer Body and Other Parties: StoriesRating: 4 out of 5 stars4/5 (821)
- Pta ResumeDocument2 pagesPta Resumeapi-669470996No ratings yet
- Stefan White, Andrew Sinclair (Auth.), John M. Hutson, Garry L. Warne, Sonia R. Grover (Eds.) - Disorders of Sex Development_ an Integrated Approach to Management-Springer-Verlag Berlin Heidelberg (20Document327 pagesStefan White, Andrew Sinclair (Auth.), John M. Hutson, Garry L. Warne, Sonia R. Grover (Eds.) - Disorders of Sex Development_ an Integrated Approach to Management-Springer-Verlag Berlin Heidelberg (20Aakanksha MehtaNo ratings yet
- Hse in Drilling OperationsDocument13 pagesHse in Drilling OperationsSamad Ali Siddiqui100% (2)
- Topical Agents and Dressings For Local Burn Wound CareDocument25 pagesTopical Agents and Dressings For Local Burn Wound CareViresh Upase Roll No 130. / 8th termNo ratings yet
- GoalSettingWorkbookFinal PDFDocument21 pagesGoalSettingWorkbookFinal PDFDato KhutsishviliNo ratings yet
- Symbols On PackegingDocument3 pagesSymbols On PackegingsakibarsNo ratings yet
- Pneumonia Care PlanDocument1 pagePneumonia Care Plantcumurphish67% (3)
- IWA Publishing - Anaerobic Reactors For Sewage Treatment - Design, Construction and Operation - 2020-01-10Document1 pageIWA Publishing - Anaerobic Reactors For Sewage Treatment - Design, Construction and Operation - 2020-01-10JOHNY ALEJANDRO GARCIA SEPULVEDANo ratings yet
- The Helping Art Clinical Nursing Who - Google SeaDocument1 pageThe Helping Art Clinical Nursing Who - Google Sea26sbn8d4p9No ratings yet
- Benign Breast DiseaseDocument26 pagesBenign Breast DiseaseMedicine 0786No ratings yet
- Methodological Literature Review 1 1Document8 pagesMethodological Literature Review 1 1api-584018105No ratings yet
- Birads PosterDocument1 pageBirads PosterGopalarathnam BalachandranNo ratings yet
- Google CardboardDocument3 pagesGoogle CardboardMartínJiménezNo ratings yet
- Lester Desjarlais Inquest Parts I and IIDocument108 pagesLester Desjarlais Inquest Parts I and IIJames Turner100% (1)
- Talisay Leaf Extract Cures Betta Fish in Less Time than Methylene BlueDocument8 pagesTalisay Leaf Extract Cures Betta Fish in Less Time than Methylene BlueMuhammad Rehan Said100% (1)
- Stetler Model EBP PosterDocument1 pageStetler Model EBP PosterEmily MNo ratings yet
- VetcareDocument18 pagesVetcareMy binNo ratings yet
- Journal Club Presentation: DR Waleed AhmadDocument30 pagesJournal Club Presentation: DR Waleed Ahmadkaram aliNo ratings yet
- Factors Affecting Social Science Teachers' Burnout in Selected State Universities in The PhilippinesDocument41 pagesFactors Affecting Social Science Teachers' Burnout in Selected State Universities in The PhilippinesAmadeus Fernando M. Pagente100% (1)
- Posthumus 2021 Competition Nutrition Practices ofDocument13 pagesPosthumus 2021 Competition Nutrition Practices ofSyazmi MohdNo ratings yet
- Newborn Care Volume 1 2020-1Document192 pagesNewborn Care Volume 1 2020-1Shyvonne PeirisNo ratings yet
- Kinds of Hazard and Risk ManagementDocument8 pagesKinds of Hazard and Risk ManagementShahid HussainNo ratings yet
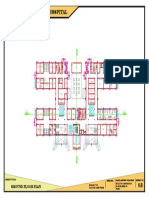
- 100-Bed General Hospital LayoutDocument1 page100-Bed General Hospital LayoutAshish chauhanNo ratings yet
- Cardiovascular NotesDocument18 pagesCardiovascular NotesCathy SantosNo ratings yet
- 1.2.1 Log 1Document3 pages1.2.1 Log 1linuspauling101100% (6)
- Kidde Fire Systems Nitrogen Engineered Systems: Design, Installation, Operation and Maintenance ManualDocument110 pagesKidde Fire Systems Nitrogen Engineered Systems: Design, Installation, Operation and Maintenance ManualYoyon HaryonoNo ratings yet
- Crodua Prioritization TableDocument10 pagesCrodua Prioritization TableThea DuoNo ratings yet
- Second Trimester Complications 2015Document64 pagesSecond Trimester Complications 2015gibreilNo ratings yet
- Function: What Is The Skeletal System?Document6 pagesFunction: What Is The Skeletal System?Mr. Christian ParabuacNo ratings yet
- Questionnaire/interview Guide Experiences of Checkpoint Volunteers During The Covid-19 PandemicDocument16 pagesQuestionnaire/interview Guide Experiences of Checkpoint Volunteers During The Covid-19 PandemicKrisna Criselda SimbreNo ratings yet