You might also like
- Manual de prácticas avanzadas para el estudio de la Microbiología ambiental de agua y sueloFrom EverandManual de prácticas avanzadas para el estudio de la Microbiología ambiental de agua y sueloRating: 4 out of 5 stars4/5 (1)
- Unidad 6 MicrobiologiaDocument52 pagesUnidad 6 MicrobiologiaIsmaelNo ratings yet
- Tecnicas Biologia MolecularDocument3 pagesTecnicas Biologia MolecularChocho CoonNo ratings yet
- Purificacion de Plamidos 2Document25 pagesPurificacion de Plamidos 2Lindas2014No ratings yet
- Conceptos Básicos de Cinética QuímicaDocument8 pagesConceptos Básicos de Cinética QuímicaNicole Del Solar LagosNo ratings yet
- Presentacion 6 Cinetica EnzimaticaDocument46 pagesPresentacion 6 Cinetica EnzimaticaE Clinton LMNo ratings yet
- Determinacion Peso Molecular Proteinas PDFDocument11 pagesDeterminacion Peso Molecular Proteinas PDFjoseNo ratings yet
- Caracteristicas de Estructuras CelularesDocument11 pagesCaracteristicas de Estructuras Celularesjose de jesus montes patricioNo ratings yet
- Lo que es un genDocument52 pagesLo que es un genAmbar GabrielaNo ratings yet
- Represion CatabolicaDocument25 pagesRepresion CatabolicaTarrillo Dávila Miguel AngelNo ratings yet
- Reacciones de La Fase Luminosa de La FotosíntesisDocument3 pagesReacciones de La Fase Luminosa de La FotosíntesisCarmen AvilaNo ratings yet
- Clonacion Del Adn RecombinanteDocument12 pagesClonacion Del Adn RecombinanteKarol MorenoNo ratings yet
- Cristalizacion de ProteinasDocument8 pagesCristalizacion de ProteinasLuis Castro100% (1)
- Factores Que Afectan El Crecimiento Microbiano.Document6 pagesFactores Que Afectan El Crecimiento Microbiano.Bryan SantoşNo ratings yet
- ElectroforesisDocument33 pagesElectroforesisJulian RodriguezNo ratings yet
- Balance EnergeticoDocument5 pagesBalance EnergeticoGus RvNo ratings yet
- Procesos Electroquimicos de Sistemas Biológicos 1Document21 pagesProcesos Electroquimicos de Sistemas Biológicos 1Gerardo Saldaña Gutierrez100% (1)
- ACTIVADOR TISULAR DE PLASMINÓGENO (t-PA) SINTESIS BIOTECNOLOGICA Y PURIFICACIÓNDocument7 pagesACTIVADOR TISULAR DE PLASMINÓGENO (t-PA) SINTESIS BIOTECNOLOGICA Y PURIFICACIÓNulises hernandezNo ratings yet
- Clonación ADN plásmidosDocument14 pagesClonación ADN plásmidosliz9207No ratings yet
- Cometabolismo - Yanza - JenifferDocument1 pageCometabolismo - Yanza - JenifferJeniffer YanzaNo ratings yet
- Diferencias clave entre la transcripción procariótica y eucarióticaDocument31 pagesDiferencias clave entre la transcripción procariótica y eucarióticaEduardo Mata GonzálezNo ratings yet
- Ensayo ElectroforesisDocument6 pagesEnsayo ElectroforesisUlises HernándezNo ratings yet
- Extraccion de ADN de BacteriasDocument9 pagesExtraccion de ADN de Bacteriassaul huarayaNo ratings yet
- Ejercicio 1 EnzimologiaDocument15 pagesEjercicio 1 EnzimologiaAndrea ChavarroNo ratings yet
- Cinetica de Reacc EnzimaticasDocument9 pagesCinetica de Reacc EnzimaticasMisato Katzuragi RitzukoNo ratings yet
- Resumen de Regulacion A Nivel DNADocument11 pagesResumen de Regulacion A Nivel DNALuis Antonio Perez AvitiaNo ratings yet
- Qué Es La Fuerza Protón MotrizDocument3 pagesQué Es La Fuerza Protón MotrizCifuentes MartinezNo ratings yet
- Reporte Ing GenéticaDocument8 pagesReporte Ing GenéticaMike BurtonNo ratings yet
- Aplicaciones de la clonación molecular: Producción de proteínas recombinantes, organismos transgénicos y terapia génicaDocument3 pagesAplicaciones de la clonación molecular: Producción de proteínas recombinantes, organismos transgénicos y terapia génicaAndrés Rodríguez MoleroNo ratings yet
- Proteinas de La CarneDocument2 pagesProteinas de La CarneJuberly DelgadoNo ratings yet
- La Modificación Covalente de La Actividad EnzimáticaDocument8 pagesLa Modificación Covalente de La Actividad EnzimáticaMaría Daisy Reyes Beltrán100% (1)
- Cinetica EnzimaticaDocument35 pagesCinetica Enzimaticajiavgr_357965222No ratings yet
- Practica No 02 MEDIOS DE CULTIVO MICROBIOLOGIADocument30 pagesPractica No 02 MEDIOS DE CULTIVO MICROBIOLOGIABelinda Mamani Mamani100% (1)
- Conservacion de Cultivos MicrobianosDocument53 pagesConservacion de Cultivos MicrobianosCesar HuallpaNo ratings yet
- Transformación Por Choque TérmicoDocument3 pagesTransformación Por Choque TérmicoPau Zanchez100% (1)
- PRACTICA 4 Determinacion de ProteinasDocument5 pagesPRACTICA 4 Determinacion de ProteinasanonidigiterNo ratings yet
- Sem 12 - Mezcla RacemicaDocument5 pagesSem 12 - Mezcla RacemicaMartin Eduardo Marchena Tirado100% (2)
- Clase 1 Biologia MolecularDocument22 pagesClase 1 Biologia MolecularZin Destino G HNo ratings yet
- Qué Es La PCRDocument4 pagesQué Es La PCRCamila ParraNo ratings yet
- Microscopio óptico: Resolución y tipos de colorantesDocument2 pagesMicroscopio óptico: Resolución y tipos de colorantesMitsuko Mendoza MajuanNo ratings yet
- Lanzaderas Malato-AspartatoDocument10 pagesLanzaderas Malato-AspartatoPaola MichaelNo ratings yet
- Ing. EnzimáticaDocument46 pagesIng. EnzimáticaArmando Montoya García100% (1)
- Respuesta de Fase AgudaDocument4 pagesRespuesta de Fase AgudaLourdes Tuanama JaraNo ratings yet
- Variantes de PCR PDFDocument21 pagesVariantes de PCR PDFraulNo ratings yet
- Transformación bacteriana y restricción de plásmidosDocument4 pagesTransformación bacteriana y restricción de plásmidosMarcela RamírezLugoNo ratings yet
- CriomicroscopíaDocument7 pagesCriomicroscopíaedisonpampaNo ratings yet
- Enzima InmovilizadaDocument8 pagesEnzima InmovilizadaYudixa ChiNo ratings yet
- Síntesis Del PeptidoglicanoDocument1 pageSíntesis Del PeptidoglicanoLissete L Agüero50% (2)
- Metabolitos Importantes en La Producción IndustrialDocument8 pagesMetabolitos Importantes en La Producción IndustrialYeferson Sebastián Santana SosaNo ratings yet
- 1mecanismos de Asimilación de SustratosDocument5 pages1mecanismos de Asimilación de Sustratosedgar gamboaNo ratings yet
- Seccic3b3n3 Curvas Temporales PDFDocument7 pagesSeccic3b3n3 Curvas Temporales PDFFernando GutiérrezNo ratings yet
- Bioquimica Tema PDFDocument20 pagesBioquimica Tema PDFLouise Clemente DoroteoNo ratings yet
- Extraccion de Dna FresaDocument12 pagesExtraccion de Dna FresaAntonioNo ratings yet
- Exposición de Ingeniería MetabólicaDocument29 pagesExposición de Ingeniería MetabólicaEduardo Fabio Apari CossioNo ratings yet
- Laboratorio Aislamiento de ADNDocument6 pagesLaboratorio Aislamiento de ADNJean FrancoNo ratings yet
- L3 DNA PlasmídicoDocument4 pagesL3 DNA PlasmídicoDanielSandovalNo ratings yet
- Aislamiento de ADN de Un Plásmido Bacteriano PDFDocument5 pagesAislamiento de ADN de Un Plásmido Bacteriano PDFMIKENo ratings yet
- Mantenimiento y Conservación de MicroorganismosDocument7 pagesMantenimiento y Conservación de Microorganismosjorge eduardoNo ratings yet
- Productos Biotecnológicos Obtenidos de Microbios ExtremófilosDocument23 pagesProductos Biotecnológicos Obtenidos de Microbios ExtremófilosPerlaZavaletaNo ratings yet
- LABORATORIO - 5 AdnDocument8 pagesLABORATORIO - 5 AdnIsabella Daza MurilloNo ratings yet
- V18n1a11 PDFDocument11 pagesV18n1a11 PDFManu RodriguezNo ratings yet
- ArnDocument11 pagesArnAngela UrbanoNo ratings yet
- ExtraccionDocument26 pagesExtraccionRichard David Aguirre BarrientosNo ratings yet
- ArnDocument11 pagesArnAngela UrbanoNo ratings yet
- 2007 0934 Remexca 7 04 00897Document14 pages2007 0934 Remexca 7 04 00897Manu RodriguezNo ratings yet
- LA GRANJA. Revista de Ciencias de La Vida 1390-3799: Issn: Sserranov@ups - Edu.ecDocument11 pagesLA GRANJA. Revista de Ciencias de La Vida 1390-3799: Issn: Sserranov@ups - Edu.ecManu RodriguezNo ratings yet
- M 011Document4 pagesM 011jordan19jvNo ratings yet
- Comparación Entre 5 Métodos para La Extracción de ADNDocument6 pagesComparación Entre 5 Métodos para La Extracción de ADNMari Carmen Juarez HernandezNo ratings yet
- ALM Estud Ustilagin 041 PDFDocument1 pageALM Estud Ustilagin 041 PDFManu RodriguezNo ratings yet
- LA GRANJA. Revista de Ciencias de La Vida 1390-3799: Issn: Sserranov@ups - Edu.ecDocument11 pagesLA GRANJA. Revista de Ciencias de La Vida 1390-3799: Issn: Sserranov@ups - Edu.ecManu RodriguezNo ratings yet
- TPAcidos NucleicosDocument7 pagesTPAcidos NucleicosbivianapazNo ratings yet
- 1089-Texto Del Artículo-3832-1-10-20130527 PDFDocument11 pages1089-Texto Del Artículo-3832-1-10-20130527 PDFManu RodriguezNo ratings yet
- INTA - CRCordoba - EEAManfredi - Grandon N. - Puesta - A - Punto - Del - Metodo - de - Extraccion - de - Adn - Con - PDFDocument1 pageINTA - CRCordoba - EEAManfredi - Grandon N. - Puesta - A - Punto - Del - Metodo - de - Extraccion - de - Adn - Con - PDFManu RodriguezNo ratings yet
- Real Time PCR HandbookDocument73 pagesReal Time PCR HandbookManu RodriguezNo ratings yet
- Un Factor de Transcripción MYB Tipo R2R3 de Trigo TaODORANT1Document14 pagesUn Factor de Transcripción MYB Tipo R2R3 de Trigo TaODORANT1Manu RodriguezNo ratings yet
- Minipreps, Tipos de Biotecnologia y Tipos de PromotoresDocument4 pagesMinipreps, Tipos de Biotecnologia y Tipos de PromotoresManu RodriguezNo ratings yet
- Actualizacion CambridgeDocument7 pagesActualizacion CambridgeManu RodriguezNo ratings yet
- Comparación Entre 5 Métodos para La Extracción de ADNDocument6 pagesComparación Entre 5 Métodos para La Extracción de ADNMari Carmen Juarez HernandezNo ratings yet
- Terapia Génica E Investigación Con Células Madre en La Legislación EspañolaDocument20 pagesTerapia Génica E Investigación Con Células Madre en La Legislación EspañolaManu RodriguezNo ratings yet
- Minipreps, Tipos de Biotecnologia y Tipos de PromotoresDocument12 pagesMinipreps, Tipos de Biotecnologia y Tipos de PromotoresManu RodriguezNo ratings yet
- Minipreps, Tipos de Biotecnologia y Tipos de PromotoresDocument12 pagesMinipreps, Tipos de Biotecnologia y Tipos de PromotoresManu RodriguezNo ratings yet
- Terapia Genica PDFDocument9 pagesTerapia Genica PDFsalvador rojasNo ratings yet
- 8870378045470Document6 pages8870378045470Manu RodriguezNo ratings yet
- 01 WatsonCrick1953Document3 pages01 WatsonCrick1953Manu Rodriguez100% (1)
- Manual de Prácticas de Bioinformática - (PG 1 - 45)Document45 pagesManual de Prácticas de Bioinformática - (PG 1 - 45)Manu Rodriguez75% (8)
- Seminario 10B - Terapia Génica PDFDocument14 pagesSeminario 10B - Terapia Génica PDFBrisa NarvaezNo ratings yet
- Actualizacion CambridgeDocument7 pagesActualizacion CambridgeManu RodriguezNo ratings yet
- Aguas de Laboratorio PDFDocument9 pagesAguas de Laboratorio PDFAntonio TrujilloNo ratings yet
- 02.3 Generalidades Sobre Al Agua Purificada PDFDocument33 pages02.3 Generalidades Sobre Al Agua Purificada PDFJ C Torres FormalabNo ratings yet
- 625 TransgenicosDocument18 pages625 TransgenicosNestor Alejandro HillarNo ratings yet
- Genetica CompletoDocument77 pagesGenetica CompletomarianoramonNo ratings yet
- Extracción DNA RNADocument35 pagesExtracción DNA RNADanny LNNo ratings yet
- ACFrOgAcY9XdinwdtrvMnyPJi AvU5whvLI4qp4tSJewMiAixffo Kmuv4H76l0 DbdeUu5ADkI 8Hgm80KRoaPLrJeDgwTgaFt52Y4tTJ-Ga1gE-h2v5LVvLfHq OD9sEuKO QBeKTteg9Yu-r3Document7 pagesACFrOgAcY9XdinwdtrvMnyPJi AvU5whvLI4qp4tSJewMiAixffo Kmuv4H76l0 DbdeUu5ADkI 8Hgm80KRoaPLrJeDgwTgaFt52Y4tTJ-Ga1gE-h2v5LVvLfHq OD9sEuKO QBeKTteg9Yu-r3Neiris Pallares CarreñoNo ratings yet
- AMINOGLUCÓSIDOSDocument6 pagesAMINOGLUCÓSIDOSgustafNo ratings yet
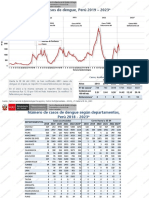
- DengueDocument55 pagesDenguejeferson100% (1)
- Boton SinapticoDocument2 pagesBoton SinapticoFranco GuzmanNo ratings yet
- Estructuras celulares: citoplasma, ribosomas, citoesqueleto, cilios y flagelosDocument16 pagesEstructuras celulares: citoplasma, ribosomas, citoesqueleto, cilios y flagelosIvanenko RivadeneiraNo ratings yet
- EXAMEN 4 y 5° AÑODocument2 pagesEXAMEN 4 y 5° AÑOBrayan CruzNo ratings yet
- Material didáctico Biología MolecularDocument46 pagesMaterial didáctico Biología MolecularJosé Luis FranzoniNo ratings yet
- Monografía de FarmacogéneticaDocument12 pagesMonografía de FarmacogéneticaFernan Calderon CabreraNo ratings yet
- Estudios in SilicoDocument4 pagesEstudios in SilicoEliseo CardenasNo ratings yet
- Practica 12 BiologiaDocument5 pagesPractica 12 BiologiaDam0% (1)
- FICHA 6 - Biomoléculas. Ácidos NucleicosDocument28 pagesFICHA 6 - Biomoléculas. Ácidos NucleicosNico CattolicaNo ratings yet
- CicloKrebsDocument5 pagesCicloKrebsk-coglyNo ratings yet
- Genoma HumanoDocument5 pagesGenoma HumanoArian Alejandra Becerra RodriguezNo ratings yet
- Do - FCS - 502 - Si - Asuc01097 - 2021Document4 pagesDo - FCS - 502 - Si - Asuc01097 - 2021JESEL FIORELA PIZARRO PARRAGANo ratings yet
- Biologia P 4Document3 pagesBiologia P 4Marta RodríguezNo ratings yet
- Seccional Guajira SegcovidDocument25 pagesSeccional Guajira SegcovidEliana Rios MoscoteNo ratings yet
- Aparato de GolgiDocument2 pagesAparato de GolgiJhonn Brandon Ventura QuisbertNo ratings yet
- Técnicas en Biología Celular y MolecularDocument10 pagesTécnicas en Biología Celular y MolecularLiz UsmeNo ratings yet
- Cromosmas. Mitosis y MeiosisDocument14 pagesCromosmas. Mitosis y MeiosisSergio Hernandez100% (1)
- Balsas LipídicasDocument4 pagesBalsas LipídicasRosa Marcela Rojas CorralesNo ratings yet
- Teoria CelularDocument9 pagesTeoria Celularlorenzo quezadaNo ratings yet
- Guía Actividad ReplicaciónDocument4 pagesGuía Actividad ReplicaciónLaura Valentina García SánchezNo ratings yet
- Papel de La Mitocondria en La Generación de AtpDocument8 pagesPapel de La Mitocondria en La Generación de AtpNicole Lopez RodriguezNo ratings yet
- A#6MSLHDocument3 pagesA#6MSLHMiguel SantiNo ratings yet
- Resultados Práctica 7Document6 pagesResultados Práctica 7Girard GarciaNo ratings yet
- METABOLISMO CARBOHIDRATOSDocument2 pagesMETABOLISMO CARBOHIDRATOSjulio sanchez camonesNo ratings yet
- TestVocacionalDocument3 pagesTestVocacionalSebastian Flores100% (4)