You might also like
- Proteomic Biology Using LC/MS: Large Scale Analysis of Cellular Dynamics and FunctionFrom EverandProteomic Biology Using LC/MS: Large Scale Analysis of Cellular Dynamics and FunctionNo ratings yet
- Proteomics - WikipediaDocument17 pagesProteomics - WikipediaP Bijoya SinghaNo ratings yet
- Basics of ProteomicsDocument7 pagesBasics of ProteomicskonndaaiahNo ratings yet
- Omics-Based On Science, Technology, and Applications OmicsDocument22 pagesOmics-Based On Science, Technology, and Applications OmicsAlex Yalew0% (1)
- Bioinformatics and ProteomicsDocument26 pagesBioinformatics and ProteomicsMohamed HasanNo ratings yet
- Omics IntroductionDocument25 pagesOmics IntroductionMarlvin PrimeNo ratings yet
- ProteomicsDocument24 pagesProteomicswatson191No ratings yet
- Biotechnology Individual AssignmentDocument10 pagesBiotechnology Individual AssignmentAlex YalewNo ratings yet
- Faculity of Natural and Computional Scince Department of Biology Biotechnology Individual AssignmentDocument10 pagesFaculity of Natural and Computional Scince Department of Biology Biotechnology Individual AssignmentAlex YalewNo ratings yet
- Bioinformatics in Cell Biology FinalDocument32 pagesBioinformatics in Cell Biology FinalBharti NawalpuriNo ratings yet
- JPP 59 5 0001Document20 pagesJPP 59 5 0001uzzi319 roony26No ratings yet
- ProteomicsDocument4 pagesProteomicsDean PhoebeNo ratings yet
- Proteo MicsDocument25 pagesProteo MicsSaptarshi GhoshNo ratings yet
- Review-2019-Proteomics Turns FunctionalDocument9 pagesReview-2019-Proteomics Turns Functionalcarlos ArozamenaNo ratings yet
- Introduction of ProteomicsDocument21 pagesIntroduction of ProteomicsMusfeera KhanNo ratings yet
- Practical Applications of Proteomics-A Technique For Large-Scale Study of Proteins: An OverviewDocument4 pagesPractical Applications of Proteomics-A Technique For Large-Scale Study of Proteins: An OverviewVishwas gargNo ratings yet
- Proteomics and GenomicsDocument23 pagesProteomics and GenomicsUma DatarNo ratings yet
- NIH Public Access: Author ManuscriptDocument14 pagesNIH Public Access: Author ManuscriptanilNo ratings yet
- Nihms449732 - Protein Analysis by Shot Gun Bottom Up ProteomicsDocument101 pagesNihms449732 - Protein Analysis by Shot Gun Bottom Up ProteomicsRoxanna LaysecaNo ratings yet
- Assignment "Proteome Analysis and Its Applications" Paper Code: BIOTECH/04/SC/28 Paper Name: Genomics and ProteomicsDocument10 pagesAssignment "Proteome Analysis and Its Applications" Paper Code: BIOTECH/04/SC/28 Paper Name: Genomics and ProteomicsLalruatdiki CNo ratings yet
- Laboratory Protocols in Fungal Biology: January 2013Document11 pagesLaboratory Protocols in Fungal Biology: January 2013lalaNo ratings yet
- System BiologyDocument16 pagesSystem BiologySubrata KunduNo ratings yet
- Novel Methodologies: Proteomic Approaches in Substance Abuse ResearchDocument12 pagesNovel Methodologies: Proteomic Approaches in Substance Abuse Researchal sharpNo ratings yet
- Comprendiendo El ProteomaDocument17 pagesComprendiendo El ProteomaWilson PolaniaNo ratings yet
- SAC Review: Omic' Technologies: Genomics, Transcriptomics, Proteomics and MetabolomicsDocument7 pagesSAC Review: Omic' Technologies: Genomics, Transcriptomics, Proteomics and MetabolomicsMiguel NavarreteNo ratings yet
- MBT31 - Proteomics NotesDocument40 pagesMBT31 - Proteomics Notesprb80730No ratings yet
- Bmir 2001 0898Document22 pagesBmir 2001 0898selena minchalaNo ratings yet
- Proteomics: The Deciphering of The Functional GenomeDocument8 pagesProteomics: The Deciphering of The Functional GenomeMaryem SafdarNo ratings yet
- Unit 1: Structural GenomicsDocument4 pagesUnit 1: Structural GenomicsLavanya ReddyNo ratings yet
- Introduction To BioinformaticsDocument10 pagesIntroduction To BioinformaticsTai Man ChanNo ratings yet
- Bio in For Ma TicsDocument26 pagesBio in For Ma Ticscsonu2209No ratings yet
- CollectionDocument8 pagesCollectionb9226276No ratings yet
- A Combined Approach For Genome Wide ProteinDocument12 pagesA Combined Approach For Genome Wide ProteinAnwar ShahNo ratings yet
- Lecture 1Document23 pagesLecture 1GopalNo ratings yet
- BioinformaticsProjects IntroductionDocument2 pagesBioinformaticsProjects IntroductionTheUnseenBeforeNo ratings yet
- Functional Proteomics To Exploit Genome Sequences: A. Donny StrosbergDocument6 pagesFunctional Proteomics To Exploit Genome Sequences: A. Donny StrosbergscribNo ratings yet
- Omics Technology: October 2010Document28 pagesOmics Technology: October 2010Janescu LucianNo ratings yet
- Single Cell Omics TechnologiesDocument3 pagesSingle Cell Omics TechnologiesRitika PathakNo ratings yet
- Overview of Proteomics: Onn Haji HashimDocument7 pagesOverview of Proteomics: Onn Haji Hashimeffak750iNo ratings yet
- Kingsmore - 2006 - Multiplexed Protein Measurement Technologies andDocument11 pagesKingsmore - 2006 - Multiplexed Protein Measurement Technologies andLaura GarciaNo ratings yet
- Vogel-2012 Transcriptome Vs ProteomeDocument13 pagesVogel-2012 Transcriptome Vs ProteomeRin ChanNo ratings yet
- 1 s2.0 S2452310017300926 MainDocument9 pages1 s2.0 S2452310017300926 MainBen DresimNo ratings yet
- Bio in For Ma TicsDocument9 pagesBio in For Ma TicsRashmi SinhaNo ratings yet
- Proteomics Tools and Applications: A. EinsteinDocument62 pagesProteomics Tools and Applications: A. EinsteintejalNo ratings yet
- Molecular Biological DetectionDocument2 pagesMolecular Biological DetectionSteven4654No ratings yet
- Cancer Diagnosis ÚingDocument10 pagesCancer Diagnosis ÚingnguyenhuulongNo ratings yet
- Introduction To NCBI ResourcesDocument39 pagesIntroduction To NCBI ResourcescgonzagaaNo ratings yet
- Computational Methods in Protein Evolution 2019Document422 pagesComputational Methods in Protein Evolution 2019DebarunAcharya100% (2)
- Proteomics IntroductionDocument39 pagesProteomics Introductionnariel67% (3)
- Chromosone AnalysisDocument234 pagesChromosone AnalysisSpataru VasileNo ratings yet
- Proteomics - Technologies and Their ApplicationsDocument15 pagesProteomics - Technologies and Their Applicationsanis k.No ratings yet
- GenomicsbookchapterDocument38 pagesGenomicsbookchapterGhinaa ZahrahNo ratings yet
- Research Papers On Protein FoldingDocument5 pagesResearch Papers On Protein Foldingshvximwhf100% (1)
- New Approaches of Protein Function Prediction from Protein Interaction NetworksFrom EverandNew Approaches of Protein Function Prediction from Protein Interaction NetworksNo ratings yet
- From Genomics To Proteomics: InsightDocument5 pagesFrom Genomics To Proteomics: Insightwadoud aggounNo ratings yet
- Proteomic TechDocument24 pagesProteomic Techbiochem instituteNo ratings yet
- Recombinant Protein Expression ThesisDocument5 pagesRecombinant Protein Expression ThesisOnlinePaperWritersCanada100% (2)
- Bioinformatics IntroDocument7 pagesBioinformatics IntroPotentflowconsultNo ratings yet
- A Full Introduction To Amino Acid Analysis Methods (Part Two)Document4 pagesA Full Introduction To Amino Acid Analysis Methods (Part Two)Steven4654No ratings yet
- Protein Analyzing Approaches and Its Application To ImmunizationDocument3 pagesProtein Analyzing Approaches and Its Application To ImmunizationSteven4654No ratings yet
- The Development History of Five Amino Acid Analysis Technologies (Part Two)Document3 pagesThe Development History of Five Amino Acid Analysis Technologies (Part Two)Steven4654No ratings yet
- Molecular Biological DetectionDocument2 pagesMolecular Biological DetectionSteven4654No ratings yet
- The Development History of Five Amino Acid Analysis Technologies (Part One)Document3 pagesThe Development History of Five Amino Acid Analysis Technologies (Part One)Steven4654No ratings yet
- Profiling On Various Kinds of GlycanDocument2 pagesProfiling On Various Kinds of GlycanSteven4654No ratings yet
- Creative Proteomics Scientists Share Precautions For Chromatin Immunoprecipitation ExperimentsDocument2 pagesCreative Proteomics Scientists Share Precautions For Chromatin Immunoprecipitation ExperimentsSteven4654No ratings yet
- Creative Proteomics Released BrdU Cell Proliferation Assay To Be Behind ResearchersDocument2 pagesCreative Proteomics Released BrdU Cell Proliferation Assay To Be Behind ResearchersSteven4654No ratings yet
- A Full Introduction To Amino Acid Analysis Methods (Part One)Document3 pagesA Full Introduction To Amino Acid Analysis Methods (Part One)Steven4654No ratings yet
- Scientists Explain The Role of DNA Methylation in Biological ActivitiesDocument2 pagesScientists Explain The Role of DNA Methylation in Biological ActivitiesSteven4654No ratings yet
- Bile Acids Play A Key Role in Regulating Intestinal Immunity and Intestinal InflammationDocument3 pagesBile Acids Play A Key Role in Regulating Intestinal Immunity and Intestinal InflammationSteven4654No ratings yet
- Β-Amino Acids Analysis ServiceDocument2 pagesΒ-Amino Acids Analysis ServiceSteven4654No ratings yet
- A Full Introduction To Amino Acid Analysis Methods (Part Three)Document3 pagesA Full Introduction To Amino Acid Analysis Methods (Part Three)Steven4654No ratings yet
- A Brief Look of CatecholamineDocument3 pagesA Brief Look of CatecholamineSteven4654No ratings yet
- Polyols Analysis ServiceDocument2 pagesPolyols Analysis ServiceSteven4654No ratings yet
- Spheroid Invasion AssayDocument2 pagesSpheroid Invasion AssaySteven4654No ratings yet
- Metabolomics BioinformaticsDocument2 pagesMetabolomics BioinformaticsSteven4654No ratings yet
- 3D Invasion AssayDocument2 pages3D Invasion AssaySteven4654No ratings yet
- Fluorescent Cell Proliferation AssayDocument1 pageFluorescent Cell Proliferation AssaySteven4654No ratings yet
- Brdu Cell ProliferationDocument1 pageBrdu Cell ProliferationSteven4654No ratings yet
- DAP Analysis ServiceDocument2 pagesDAP Analysis ServiceSteven4654No ratings yet
- Respiratory Quinones AnalysisDocument2 pagesRespiratory Quinones AnalysisSteven4654No ratings yet
- MTS AssayDocument2 pagesMTS AssaySteven4654No ratings yet
- Bioinformatics ServicesDocument1 pageBioinformatics ServicesSteven4654No ratings yet
- Polyols Analysis ServiceDocument2 pagesPolyols Analysis ServiceSteven4654No ratings yet
- Glycosaminoglycans Analysis ServiceDocument2 pagesGlycosaminoglycans Analysis ServiceSteven4654No ratings yet
- Ubiquinone Analysis ServiceDocument2 pagesUbiquinone Analysis ServiceSteven4654No ratings yet
- Flavin SDocument4 pagesFlavin SSteven4654No ratings yet
- SAM and SAH Analysis ServiceDocument2 pagesSAM and SAH Analysis ServiceSteven4654No ratings yet
- Selina Solutions For Class 9 Biology Chapter 2 Cell The Unit of LifeDocument15 pagesSelina Solutions For Class 9 Biology Chapter 2 Cell The Unit of LifePrashun PriyadarshiNo ratings yet
- Tule Elk in Point ReyesDocument4 pagesTule Elk in Point ReyesPointReyesNo ratings yet
- Ch8 (C)Document28 pagesCh8 (C)fkjujNo ratings yet
- Hamsters - A General DescriptionDocument5 pagesHamsters - A General Descriptiondulce crissNo ratings yet
- Recent DevelopmentsDocument295 pagesRecent DevelopmentsGabriel PinheiroNo ratings yet
- CAPE Bio Mark SchemeDocument4 pagesCAPE Bio Mark Schemeron97150% (2)
- 3-MS Examples PDFDocument59 pages3-MS Examples PDFlexuanminhk54lochoaNo ratings yet
- Sri Roth 2000Document11 pagesSri Roth 2000ottoojuniiorNo ratings yet
- Social Institutions - Kinship, Family&Marriage PDFDocument47 pagesSocial Institutions - Kinship, Family&Marriage PDFAbhijith KNo ratings yet
- Determination of Sensitive Proteins in Beer by Nephelometry - Submitted On Behalf of The Analysis Committee of The European Brewery ConventionDocument4 pagesDetermination of Sensitive Proteins in Beer by Nephelometry - Submitted On Behalf of The Analysis Committee of The European Brewery ConventionChí HữuNo ratings yet
- Chapter 1Document4 pagesChapter 1Nikoleta RudnikNo ratings yet
- Kami Export - Omarion Fladger - Pedigree Genetics ProblemsDocument2 pagesKami Export - Omarion Fladger - Pedigree Genetics ProblemsOmarion FladgerNo ratings yet
- Cham PowerpointDocument19 pagesCham PowerpointElizabeth GenotivaNo ratings yet
- Capsicum Annuum L., Commonly Known As Hot Pepper or Chilli Is ADocument11 pagesCapsicum Annuum L., Commonly Known As Hot Pepper or Chilli Is Ajavedsaqi100% (1)
- Killing of Kangaroos For Meat and SkinDocument34 pagesKilling of Kangaroos For Meat and SkinVegan FutureNo ratings yet
- (Springer Series in Wood Science) W. E. Hillis (auth.), John W. Rowe (eds.)-Natural Products of Woody Plants_ Chemicals Extraneous to the Lignocellulosic Cell Wall-Springer-Verlag Berlin Heidelberg (1.pdfDocument1,274 pages(Springer Series in Wood Science) W. E. Hillis (auth.), John W. Rowe (eds.)-Natural Products of Woody Plants_ Chemicals Extraneous to the Lignocellulosic Cell Wall-Springer-Verlag Berlin Heidelberg (1.pdfArmandoNo ratings yet
- Science of The Total Environment: Megan Carve Graeme Allinson, Dayanthi Nugegoda, Jeff ShimetaDocument16 pagesScience of The Total Environment: Megan Carve Graeme Allinson, Dayanthi Nugegoda, Jeff ShimetaFABIANA PASSAMANINo ratings yet
- An Examination and Critique of Current Methods To Determine Exercise IntensityDocument28 pagesAn Examination and Critique of Current Methods To Determine Exercise IntensityPabloAñonNo ratings yet
- Biosensors For Environmental ApplicationsDocument15 pagesBiosensors For Environmental ApplicationsBenjamin HonorioNo ratings yet
- Nominalization (Exercise)Document2 pagesNominalization (Exercise)KryzQuinonesNo ratings yet
- Human Genome Editing: Science, Ethics, and GovernanceDocument261 pagesHuman Genome Editing: Science, Ethics, and GovernanceAnonymous 6VKlaeigViNo ratings yet
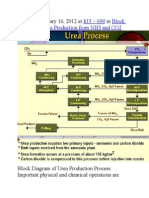
- Published January 16, 2012 at In: 813 × 699 Block Diagram of Urea Production From NH3 and CO2Document9 pagesPublished January 16, 2012 at In: 813 × 699 Block Diagram of Urea Production From NH3 and CO2himanshuchawla654No ratings yet
- Principles of Inheritance and Variations: By: Dr. Anand ManiDocument113 pagesPrinciples of Inheritance and Variations: By: Dr. Anand ManiIndu YadavNo ratings yet
- Guideline For Prevention of Surgical Wound InfectionsDocument14 pagesGuideline For Prevention of Surgical Wound InfectionsosaqerNo ratings yet
- Third Stage Begg MechanotherapyDocument32 pagesThird Stage Begg MechanotherapyNaziya ShaikNo ratings yet
- Ingles Medico Ii - TP 5Document3 pagesIngles Medico Ii - TP 5Rodrigo Anelli De OliveiraNo ratings yet
- Short Note Biology Form 5-Chapter 3 Coordination and ResponseDocument6 pagesShort Note Biology Form 5-Chapter 3 Coordination and Responsesalamah_sabri75% (4)
- Staar Eoc 2016test Bio F 7Document39 pagesStaar Eoc 2016test Bio F 7api-293216402No ratings yet
- A Review On Potential Toxicity of Artificial Sweetners Vs Safety of SteviaDocument13 pagesA Review On Potential Toxicity of Artificial Sweetners Vs Safety of SteviaAlexander DeckerNo ratings yet
- Johnson Jerry Alan Chinese Medical Qigong Therapy Vol 5-141-160Document20 pagesJohnson Jerry Alan Chinese Medical Qigong Therapy Vol 5-141-160toanbauNo ratings yet