You might also like
- Molecular Docking Shifting Paradigms in Drug DiscoDocument23 pagesMolecular Docking Shifting Paradigms in Drug Discorashamsd9No ratings yet
- Pharmacology in Drug Discovery: Understanding Drug ResponseFrom EverandPharmacology in Drug Discovery: Understanding Drug ResponseNo ratings yet
- 06 - Chapter 1Document37 pages06 - Chapter 1Vikash KushwahaNo ratings yet
- Computer Aided Drug Design The Most Fundamental Goal Is To Predict Whether A Given Molecule Will Bind To A Target and If So How StronglyDocument6 pagesComputer Aided Drug Design The Most Fundamental Goal Is To Predict Whether A Given Molecule Will Bind To A Target and If So How StronglyAlexander DeckerNo ratings yet
- Practise School - ChemistryDocument64 pagesPractise School - ChemistrySonakshi BhatiaNo ratings yet
- Term Paper: Various Techniques in Drug SynthesisDocument14 pagesTerm Paper: Various Techniques in Drug SynthesisUmair MazharNo ratings yet
- ComputerDocument2 pagesComputerAmina AshrafNo ratings yet
- Drug Discovery StrategiesDocument5 pagesDrug Discovery Strategiesminad53929 dineroacomNo ratings yet
- Molecular Docking PaperDocument24 pagesMolecular Docking PaperChristian CastroNo ratings yet
- En Wikipedia OrgDocument3 pagesEn Wikipedia OrgMilthon PerezNo ratings yet
- Recent Advances in Rational Drug Design: Paramjeet Kaur, Akashdeep, MukulDocument35 pagesRecent Advances in Rational Drug Design: Paramjeet Kaur, Akashdeep, MukulParamjeet Kaur Paramedical SciencesNo ratings yet
- Link Prediction Drug DiseaseDocument21 pagesLink Prediction Drug Disease20102076No ratings yet
- Virtual Screening ReportDocument4 pagesVirtual Screening ReportManasa UpadhyayaNo ratings yet
- Deep Learning Based Drug Metabolites PredictionDocument11 pagesDeep Learning Based Drug Metabolites PredictionMony MontelongoNo ratings yet
- Ligand-Target Prediction by Structural Network Biology Using NannolyzeDocument19 pagesLigand-Target Prediction by Structural Network Biology Using Nannolyze1pisco1No ratings yet
- Insilico 3Document12 pagesInsilico 3Naveen Virendra SinghNo ratings yet
- A Review On Molecular Docking and Its ApplicationDocument13 pagesA Review On Molecular Docking and Its ApplicationIJAR JOURNALNo ratings yet
- Agoni 2020Document11 pagesAgoni 2020Daniel Bustos GuajardoNo ratings yet
- In Silico Pharmacology For Drug Discovery: Applications To Targets and BeyondDocument17 pagesIn Silico Pharmacology For Drug Discovery: Applications To Targets and BeyondAw Yong Yi XiangNo ratings yet
- Introduction To Computer Aided Drug DesignDocument85 pagesIntroduction To Computer Aided Drug DesignHanumant Suryawanshi81% (21)
- 2016 J Cheminform 8 14Document16 pages2016 J Cheminform 8 14mbrylinskiNo ratings yet
- SBDD An OverviewDocument16 pagesSBDD An OverviewAnkita SinghNo ratings yet
- MolecularDockingStructureBasedDrugDesignStrategies PDFDocument38 pagesMolecularDockingStructureBasedDrugDesignStrategies PDFIvt Morales100% (1)
- Drug 4Document15 pagesDrug 4Salem Optimus Technocrates India Private LimitedNo ratings yet
- Machine-Learning Methods For Ligand-Protein Molecular DockingDocument17 pagesMachine-Learning Methods For Ligand-Protein Molecular DockingInes MejriNo ratings yet
- BasedpairedDocument34 pagesBasedpairedRicardo RomeroNo ratings yet
- Computer Aided Drug DesignDocument40 pagesComputer Aided Drug DesignHanumant Suryawanshi0% (1)
- Review Article Introduction To Chemical Proteomics For Drug Discovery and DevelopmentDocument9 pagesReview Article Introduction To Chemical Proteomics For Drug Discovery and DevelopmentPaviliuc RalucaNo ratings yet
- Computational Methods For Prediction of Drug LikenessDocument10 pagesComputational Methods For Prediction of Drug LikenesssciencystuffNo ratings yet
- "Probability and Statistics" " (ASSIGNMENT #2) "Document24 pages"Probability and Statistics" " (ASSIGNMENT #2) "ahmedNo ratings yet
- N Asmita Bandyopadhyay 2233645 - Cia IDocument10 pagesN Asmita Bandyopadhyay 2233645 - Cia IN ASMITA BANDYOPADHYAY 2233645No ratings yet
- Drug Designing 1Document2 pagesDrug Designing 1ShreyaChakladarNo ratings yet
- Acharya Et Al, 2011 - Recent Advances in Ligand-Based Drug Design Relevance and Utility of The Conformationally Sampled Pharmacophore ApproachDocument26 pagesAcharya Et Al, 2011 - Recent Advances in Ligand-Based Drug Design Relevance and Utility of The Conformationally Sampled Pharmacophore ApproachDiana López LópezNo ratings yet
- s12859 017 1638 4 PDFDocument10 pagess12859 017 1638 4 PDFfkamaliyahNo ratings yet
- 29.keshri Kishore Jha Ravi TripathiDocument16 pages29.keshri Kishore Jha Ravi TripathiTukai KulkarniNo ratings yet
- Bioinformatics: Original PaperDocument8 pagesBioinformatics: Original PaperfkamaliyahNo ratings yet
- Brian K Shoichet, Susan L Mcgovern, Binqing Wei and John J IrwinDocument8 pagesBrian K Shoichet, Susan L Mcgovern, Binqing Wei and John J Irwinsrikalyani2k9No ratings yet
- Genes 12 00025 v2Document12 pagesGenes 12 00025 v2王汉扬No ratings yet
- De Vivo, M. Et Al. Role of Molecular Dynamics and Related Methods in Drug Discovery. J. Med. Chem. 59, 4035-4061 (2016) .Document27 pagesDe Vivo, M. Et Al. Role of Molecular Dynamics and Related Methods in Drug Discovery. J. Med. Chem. 59, 4035-4061 (2016) .Thales FreireNo ratings yet
- Drug-Target Interaction Prediction in Drug Repositioning Based On Deep Semi-Supervised LearningDocument12 pagesDrug-Target Interaction Prediction in Drug Repositioning Based On Deep Semi-Supervised LearningBilal AhmadNo ratings yet
- 9&8 RefDocument7 pages9&8 RefEswaran RameshNo ratings yet
- Molecular Docking TheoryDocument21 pagesMolecular Docking Theoryolivia6669No ratings yet
- Role of Computer-Aided Drug Design in Modern Drug DiscoveryDocument16 pagesRole of Computer-Aided Drug Design in Modern Drug DiscoverySourav MajumdarNo ratings yet
- Protein Interaction Mol DockingDocument49 pagesProtein Interaction Mol DockingKhuwaylaNo ratings yet
- Review On Computational Bioinformatics and Molecular Modelling Novel Tool For Drug DiscoveryDocument6 pagesReview On Computational Bioinformatics and Molecular Modelling Novel Tool For Drug DiscoveryEditor IJTSRDNo ratings yet
- Bioinformatics Project On Drug Discovery and Drug DesigningDocument10 pagesBioinformatics Project On Drug Discovery and Drug DesigningJatin AroraNo ratings yet
- Affinity Selection-Mass Spectrometry Screening Techniques For Small Molecule Drug DiscoveryDocument9 pagesAffinity Selection-Mass Spectrometry Screening Techniques For Small Molecule Drug DiscoveryFábio Teixeira da SilvaNo ratings yet
- Drug DesignDocument11 pagesDrug Designnevelle4667No ratings yet
- Aps 2012109Document10 pagesAps 2012109Stym ÅsàtïNo ratings yet
- Review 1-5 PDFDocument5 pagesReview 1-5 PDFEditor IjprtNo ratings yet
- Biology Project On Ai in MedicineDocument10 pagesBiology Project On Ai in Medicinejapmansingh45678No ratings yet
- Bioiphysical Methods in Early Drug DiscoveryDocument20 pagesBioiphysical Methods in Early Drug Discoveryrenz montallanaNo ratings yet
- A Structure-Based Drug Discovery Paradigm: Molecular SciencesDocument18 pagesA Structure-Based Drug Discovery Paradigm: Molecular SciencesfreeretdocsNo ratings yet
- Deep Learning With Feature Embedding For Compound-Protein Interaction PredictionDocument21 pagesDeep Learning With Feature Embedding For Compound-Protein Interaction Predictionwangxiangwen0201No ratings yet
- Prediction of Drug-Target Interactions and Drug Repositioning Via Network-Based InferenceDocument12 pagesPrediction of Drug-Target Interactions and Drug Repositioning Via Network-Based Inferencesamuel chenNo ratings yet
- Srep 24245Document14 pagesSrep 24245TakashiSuzukiNo ratings yet
- A Benchmark Driven Guide To Binding SiteDocument50 pagesA Benchmark Driven Guide To Binding SiteAlexNo ratings yet
- Computer-Aided Drug DesignDocument6 pagesComputer-Aided Drug DesignAnand Barapatre100% (1)
- Pharmaceutics: Ionic Channels As Targets For Drug Design: A Review On Computational MethodsDocument22 pagesPharmaceutics: Ionic Channels As Targets For Drug Design: A Review On Computational MethodsShatanik MukherjeeNo ratings yet
- chetan-mukul-ICCM21 PPT-final-part-2 PDFDocument15 pageschetan-mukul-ICCM21 PPT-final-part-2 PDFMukul ShuklaNo ratings yet
- M2 Periodic Table PDFDocument30 pagesM2 Periodic Table PDFMukul ShuklaNo ratings yet
- chetan-mukul-ICCM21 PPT-final-part-2 PDFDocument15 pageschetan-mukul-ICCM21 PPT-final-part-2 PDFMukul ShuklaNo ratings yet
- Chetan Mukul ICCM21 PPT Final Part 1Document18 pagesChetan Mukul ICCM21 PPT Final Part 1Mukul ShuklaNo ratings yet
- Chetan Mukul ICCM21 PPT Final Part 1Document18 pagesChetan Mukul ICCM21 PPT Final Part 1Mukul ShuklaNo ratings yet
- Chetan Mukul ICCM21 PPT Final Part 1Document18 pagesChetan Mukul ICCM21 PPT Final Part 1Mukul ShuklaNo ratings yet
- Saiche Paper Pilusa V19 n1 2014 PP 22 30Document9 pagesSaiche Paper Pilusa V19 n1 2014 PP 22 30Mukul ShuklaNo ratings yet
- Know All About JEE AdvancedDocument20 pagesKnow All About JEE AdvancedMukul ShuklaNo ratings yet
- Jcam Naresh Mukul Awjm Fem Paper 2012Document11 pagesJcam Naresh Mukul Awjm Fem Paper 2012Mukul ShuklaNo ratings yet
- Ajay Paper Ijstm 021211Document12 pagesAjay Paper Ijstm 021211Mukul ShuklaNo ratings yet
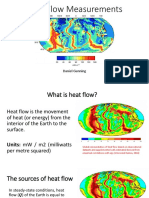
- Heat FlowDocument22 pagesHeat FlowIshita MongaNo ratings yet
- Photon noise: τ σ = σ hf n wattsDocument24 pagesPhoton noise: τ σ = σ hf n wattssiddhanta69No ratings yet
- Chemistry Project - BilalDocument6 pagesChemistry Project - Bilalmetrotigers377No ratings yet
- Rubber Process Oils NewDocument4 pagesRubber Process Oils NewVaradrajan jothiNo ratings yet
- Molecular Diagnostics 4ADocument6 pagesMolecular Diagnostics 4APaul LesterNo ratings yet
- Acoustics 4Document24 pagesAcoustics 4That GuyNo ratings yet
- Vision Filter CatalogueDocument9 pagesVision Filter CataloguevisionfilterNo ratings yet
- 5 Chapter 4Document15 pages5 Chapter 4azizNo ratings yet
- J Chroma 2017 12 056Document9 pagesJ Chroma 2017 12 056AnisaNo ratings yet
- School Base Assessment-1Document48 pagesSchool Base Assessment-1Marissa FreemanNo ratings yet
- Topworx ValvetopDocument24 pagesTopworx ValvetopaleNo ratings yet
- Lecture 1Document23 pagesLecture 1meku44No ratings yet
- What Is Heat TransferDocument93 pagesWhat Is Heat TransferPradeep RavichandranNo ratings yet
- Dynamic Model For Nutrient Uptake by Tomato Plant in HydroponicsDocument46 pagesDynamic Model For Nutrient Uptake by Tomato Plant in HydroponicsAmmarsalNo ratings yet
- NBP Planner - NEET-2022Document21 pagesNBP Planner - NEET-2022Mruthyun120% (1)
- Chemical Composition of Callisia Fragrans Juice 1. Phenolic CompoundsDocument2 pagesChemical Composition of Callisia Fragrans Juice 1. Phenolic CompoundsLeTienDungNo ratings yet
- PVD CVD WNP PDFDocument71 pagesPVD CVD WNP PDFApresiasi teknik 2018No ratings yet
- Isrm SM Petrographic Description - 1978 PDFDocument3 pagesIsrm SM Petrographic Description - 1978 PDFEvandro Santiago100% (2)
- Scheme of Work Science Stage 8Document90 pagesScheme of Work Science Stage 8Arjun SrinivasanNo ratings yet
- TEMP ABB Guide PDFDocument323 pagesTEMP ABB Guide PDFjavad4531No ratings yet
- Impact of Heterogeneous Cavities On The Electrical Constraints in The Insulation of High-Voltage CablesDocument9 pagesImpact of Heterogeneous Cavities On The Electrical Constraints in The Insulation of High-Voltage CablesOlga OliveiraNo ratings yet
- SAE J113-1998 ScanDocument3 pagesSAE J113-1998 ScanMarcos RosenbergNo ratings yet
- Summative Test #4 Science 6Document2 pagesSummative Test #4 Science 6chona redillasNo ratings yet
- Department of Chemical Engineering: National Institute of Technology, RaipurDocument4 pagesDepartment of Chemical Engineering: National Institute of Technology, RaipurHimanshu SinghNo ratings yet
- Analyze The Cases and Answer The QuestionsDocument31 pagesAnalyze The Cases and Answer The QuestionsJohn Lloyd PedresoNo ratings yet
- Exp 5 and 6 Lab Report PDFDocument10 pagesExp 5 and 6 Lab Report PDFIsabel Joice EnriquezNo ratings yet
- The Effect of Monosaccharides Versus Disaccharides On The Rate of CO ProductionDocument4 pagesThe Effect of Monosaccharides Versus Disaccharides On The Rate of CO ProductionRyan LamNo ratings yet
- Kukatpally, Hyderabad - 500072.: Narayana Junior College - Hyd - KPBG - 1580 Junior Intermediate Cbse DATE - 11/04/2023Document2 pagesKukatpally, Hyderabad - 500072.: Narayana Junior College - Hyd - KPBG - 1580 Junior Intermediate Cbse DATE - 11/04/2023StandbyNo ratings yet
- Revision SPM 2018 Paper 2Document70 pagesRevision SPM 2018 Paper 2Azie Nurul Akhtar75% (4)
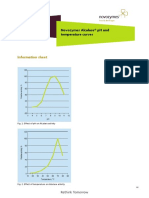
- Alcalase 2.5 L - PH and TemperatureDocument2 pagesAlcalase 2.5 L - PH and TemperatureAída MorenoNo ratings yet
- The Periodic Table of Elements - Post-Transition Metals, Metalloids and Nonmetals | Children's Chemistry BookFrom EverandThe Periodic Table of Elements - Post-Transition Metals, Metalloids and Nonmetals | Children's Chemistry BookNo ratings yet
- Physical and Chemical Equilibrium for Chemical EngineersFrom EverandPhysical and Chemical Equilibrium for Chemical EngineersRating: 5 out of 5 stars5/5 (1)
- Process Plant Equipment: Operation, Control, and ReliabilityFrom EverandProcess Plant Equipment: Operation, Control, and ReliabilityRating: 5 out of 5 stars5/5 (1)
- Guidelines for the Management of Change for Process SafetyFrom EverandGuidelines for the Management of Change for Process SafetyNo ratings yet
- Nuclear Energy in the 21st Century: World Nuclear University PressFrom EverandNuclear Energy in the 21st Century: World Nuclear University PressRating: 4.5 out of 5 stars4.5/5 (3)
- Process Steam Systems: A Practical Guide for Operators, Maintainers, and DesignersFrom EverandProcess Steam Systems: A Practical Guide for Operators, Maintainers, and DesignersNo ratings yet
- The Periodic Table of Elements - Alkali Metals, Alkaline Earth Metals and Transition Metals | Children's Chemistry BookFrom EverandThe Periodic Table of Elements - Alkali Metals, Alkaline Earth Metals and Transition Metals | Children's Chemistry BookNo ratings yet
- Well Control for Completions and InterventionsFrom EverandWell Control for Completions and InterventionsRating: 4 out of 5 stars4/5 (10)
- Gas-Liquid And Liquid-Liquid SeparatorsFrom EverandGas-Liquid And Liquid-Liquid SeparatorsRating: 3.5 out of 5 stars3.5/5 (3)
- Phase Equilibria in Chemical EngineeringFrom EverandPhase Equilibria in Chemical EngineeringRating: 4 out of 5 stars4/5 (11)
- An Introduction to the Periodic Table of Elements : Chemistry Textbook Grade 8 | Children's Chemistry BooksFrom EverandAn Introduction to the Periodic Table of Elements : Chemistry Textbook Grade 8 | Children's Chemistry BooksRating: 5 out of 5 stars5/5 (1)
- Guidelines for Chemical Process Quantitative Risk AnalysisFrom EverandGuidelines for Chemical Process Quantitative Risk AnalysisRating: 5 out of 5 stars5/5 (1)
- Pharmaceutical Blending and MixingFrom EverandPharmaceutical Blending and MixingP. J. CullenRating: 5 out of 5 stars5/5 (1)
- Piping and Pipeline Calculations Manual: Construction, Design Fabrication and ExaminationFrom EverandPiping and Pipeline Calculations Manual: Construction, Design Fabrication and ExaminationRating: 4 out of 5 stars4/5 (18)
- Operational Excellence: Journey to Creating Sustainable ValueFrom EverandOperational Excellence: Journey to Creating Sustainable ValueNo ratings yet
- Guidelines for Engineering Design for Process SafetyFrom EverandGuidelines for Engineering Design for Process SafetyNo ratings yet
- Sodium Bicarbonate: Nature's Unique First Aid RemedyFrom EverandSodium Bicarbonate: Nature's Unique First Aid RemedyRating: 5 out of 5 stars5/5 (21)
- Understanding Process Equipment for Operators and EngineersFrom EverandUnderstanding Process Equipment for Operators and EngineersRating: 4.5 out of 5 stars4.5/5 (3)
- The HAZOP Leader's Handbook: How to Plan and Conduct Successful HAZOP StudiesFrom EverandThe HAZOP Leader's Handbook: How to Plan and Conduct Successful HAZOP StudiesNo ratings yet
- Chemical Process Safety: Learning from Case HistoriesFrom EverandChemical Process Safety: Learning from Case HistoriesRating: 4 out of 5 stars4/5 (14)
- Functional Safety from Scratch: A Practical Guide to Process Industry ApplicationsFrom EverandFunctional Safety from Scratch: A Practical Guide to Process Industry ApplicationsNo ratings yet