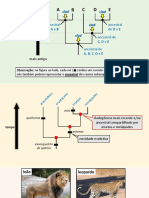
You might also like
- Filogenia e CladogramasDocument48 pagesFilogenia e CladogramasamidoadelinoNo ratings yet
- Nocoes Sistematica Filogenetica PDFDocument3 pagesNocoes Sistematica Filogenetica PDFjogoalberNo ratings yet
- Atividade CladogramaDocument6 pagesAtividade CladogramaArthurNo ratings yet
- 3AULA - 2 - Sistema de Classificação CladísticaDocument24 pages3AULA - 2 - Sistema de Classificação CladísticaAlisson Roehrs100% (1)
- Taxonomia Sistematica FilogeneticaDocument38 pagesTaxonomia Sistematica FilogeneticaTxai Evan Brandao100% (1)
- Diversidade Das AngiospermasDocument111 pagesDiversidade Das AngiospermasJnani ArlogNo ratings yet
- Estudo Dirigido de Diversidade BiológicaDocument12 pagesEstudo Dirigido de Diversidade BiológicaAline MartinsNo ratings yet
- Padrões e Processos Ecológicos e Evolutivos em Escala Regional - 2009 PDFDocument12 pagesPadrões e Processos Ecológicos e Evolutivos em Escala Regional - 2009 PDFGaby F De DeusNo ratings yet
- Aula (07-11) - Teias AlimentaresDocument61 pagesAula (07-11) - Teias AlimentaresGermano Henrique Costa BarrilliNo ratings yet
- Classificação BiologicaDocument61 pagesClassificação BiologicaIsa Pacheco100% (1)
- Sist Classificação 2023Document19 pagesSist Classificação 2023Átila Borges VianaNo ratings yet
- Introdução A MetazoaDocument24 pagesIntrodução A MetazoaJeane AlmeidaNo ratings yet
- Extinção e IrradiaçãoDocument77 pagesExtinção e IrradiaçãoThiago MartinsNo ratings yet
- BriófitasDocument10 pagesBriófitasFrancisco SallasNo ratings yet
- Aula EvoluçãoDocument26 pagesAula Evoluçãoantonio leaoNo ratings yet
- SLIDES Da Aula - Bio 3 - Semana 2 - MED - Caio GadelDocument12 pagesSLIDES Da Aula - Bio 3 - Semana 2 - MED - Caio Gadelbo yyNo ratings yet
- Aula 5 - Filogenias, Cladogramas e Árvores FilogenéticaDocument14 pagesAula 5 - Filogenias, Cladogramas e Árvores FilogenéticaAndré LucasNo ratings yet
- Aula 13 - Biodiversidade PDFDocument56 pagesAula 13 - Biodiversidade PDFVitor VitalNo ratings yet
- Apostila - Entomologia - Basica (Taxonomia, Identificação, Nomenclatura de Especies)Document50 pagesApostila - Entomologia - Basica (Taxonomia, Identificação, Nomenclatura de Especies)antonioandreoNo ratings yet
- HEREDOGRAMASDocument4 pagesHEREDOGRAMASThiago RockenbachNo ratings yet
- Aula 4.2 - Historia de Vida PDFDocument61 pagesAula 4.2 - Historia de Vida PDFPhabloDiasNo ratings yet
- Origem e Evolução Dos InsetosDocument41 pagesOrigem e Evolução Dos InsetosAdenomar Neves de Carvalho67% (3)
- Ecologia VegetalDocument7 pagesEcologia VegetalDan PaskinNo ratings yet
- O Neo-DarwinismoDocument57 pagesO Neo-Darwinismodavidpais-ovar0% (1)
- Interações EcologicasDocument29 pagesInterações EcologicasArianne FabresNo ratings yet
- Apostila 1 de Introdução Á BotânicaDocument100 pagesApostila 1 de Introdução Á Botânicavegano navegandoNo ratings yet
- Respostas A Economia Da Natureza Capitulo 6Document2 pagesRespostas A Economia Da Natureza Capitulo 6Ozimar Froes100% (4)
- Várias Teorias Evolutivas SurgiramDocument3 pagesVárias Teorias Evolutivas SurgiramPreis1No ratings yet
- 5.-CnidáriosDocument7 pages5.-CnidáriosFernando FreitasNo ratings yet
- 1 4924855796493189261Document111 pages1 4924855796493189261Marcelo ClaroNo ratings yet
- Especiação e FilogeniaDocument3 pagesEspeciação e FilogeniaVanessa NogueiraNo ratings yet
- Evolução Teorias e Evidências - Ex - OkDocument15 pagesEvolução Teorias e Evidências - Ex - OkAurilene SouzaNo ratings yet
- Anatomia Da Raiz PDFDocument45 pagesAnatomia Da Raiz PDFAnonymous rUzMFGK8qGNo ratings yet
- Classificação BotânicaDocument3 pagesClassificação Botânicamatheus11alvesmNo ratings yet
- Biologia - Idéias EvolucionistasDocument10 pagesBiologia - Idéias Evolucionistasediab2008100% (4)
- Ex - A1 - Aula 05 - Origem Da Vida e Evolução PDFDocument4 pagesEx - A1 - Aula 05 - Origem Da Vida e Evolução PDFjoaoNo ratings yet
- Cap 7 - Evol Plantas - Sistemática Vegetal Udd Et Al (Compressed)Document32 pagesCap 7 - Evol Plantas - Sistemática Vegetal Udd Et Al (Compressed)GioNo ratings yet
- 08 - Evolucionismo e FixismoDocument30 pages08 - Evolucionismo e FixismoMartaNo ratings yet
- Atividade Botânica I 2022Document5 pagesAtividade Botânica I 2022Michelle Soares Pereira100% (1)
- Classificação Dos Seres VivosDocument5 pagesClassificação Dos Seres VivosCauã TVNo ratings yet
- Exercícios GenéticaDocument6 pagesExercícios GenéticaLucimara FernandesNo ratings yet
- Glossário de Termos BotânicosDocument3 pagesGlossário de Termos BotânicosFrancisco BezerraNo ratings yet
- AtividadeDocument5 pagesAtividadeThaila MaiuryNo ratings yet
- Introdução A Genética 1 Lei de Mendel Outubro - 104324Document39 pagesIntrodução A Genética 1 Lei de Mendel Outubro - 104324Anne Lara AlvesNo ratings yet
- Resumo AngiospermasDocument3 pagesResumo AngiospermasEwerthon GomesNo ratings yet
- Aula 9. FotoperiodismoDocument27 pagesAula 9. FotoperiodismoDanielly C. Schupchek100% (1)
- Resumo DSV Aulas 1-32 PDFDocument58 pagesResumo DSV Aulas 1-32 PDFFDLNo ratings yet
- Evolucao BiologicaDocument8 pagesEvolucao BiologicaMargarida Passanha100% (1)
- Apostila ArthropodaDocument35 pagesApostila Arthropodawendellupton8035No ratings yet
- Angiospermas ClassificaçãoDocument32 pagesAngiospermas ClassificaçãoRhuan CarvalhoNo ratings yet
- 1 PDFDocument102 pages1 PDFKaylaneNo ratings yet
- Fungos - Sonia LopesDocument7 pagesFungos - Sonia LopesFátima FafaNo ratings yet
- Aula 04Document16 pagesAula 04GIORDANNO2299No ratings yet
- ArtropodesDocument5 pagesArtropodesSebastião LopesNo ratings yet
- Classificação Dos Seres Vivos TextoDocument6 pagesClassificação Dos Seres Vivos TextoPatrícia Alexandra da Silva Paz100% (1)
- AcidosexDocument1 pageAcidosexMaria Cristina Lima RosaNo ratings yet
- BiologiaMolecular Texto04 (8) FinalDocument23 pagesBiologiaMolecular Texto04 (8) Finaljoao victor0% (2)
- Bases Moleculares - AulaDocument24 pagesBases Moleculares - AulaEdu OMNo ratings yet
- Genética - Wikipédia, A Enciclopédia LivreDocument44 pagesGenética - Wikipédia, A Enciclopédia Livresardio dos santosNo ratings yet
- ADRENOCORTICÓIDESDocument38 pagesADRENOCORTICÓIDESsidsousaNo ratings yet
- Alterações CromossómicasDocument2 pagesAlterações CromossómicasRayane KelliNo ratings yet
- Listas de Exercícios Tutores para Seu Enem 003Document3 pagesListas de Exercícios Tutores para Seu Enem 003fernandogastNo ratings yet
- Lista de Exercícios I FBM 2023Document1 pageLista de Exercícios I FBM 2023giovannar.belatableNo ratings yet
- Estudo Da CelulaDocument3 pagesEstudo Da CelulaemarianoNo ratings yet
- Quimiot Aula 4 Agentes Antimetabolitos 2019Document12 pagesQuimiot Aula 4 Agentes Antimetabolitos 2019Samuel JosexNo ratings yet
- Modelo de Apresentação Anhanguera CorrigidoDocument14 pagesModelo de Apresentação Anhanguera CorrigidoLuana GabrielNo ratings yet
- Paródia Baile de OrganelaDocument1 pageParódia Baile de OrganelaRenan De Jesus Pontes Camargo75% (8)
- Respiração Aeróbica BioDocument14 pagesRespiração Aeróbica BioLeonor MaiaNo ratings yet
- Aplicações Da Genética Na SociedadeDocument8 pagesAplicações Da Genética Na SociedadeAna Sofia SantosNo ratings yet
- Variações Das Leis de Mendel PDFDocument5 pagesVariações Das Leis de Mendel PDFmarceloNo ratings yet
- Necrose CaseosaDocument2 pagesNecrose CaseosaLara PereiraNo ratings yet
- Aula 10 Sintese ProteicaDocument43 pagesAula 10 Sintese Proteicaapi-19738142100% (1)
- Fisiopatologia e Farmacologia Aplicada À NutriçãoDocument5 pagesFisiopatologia e Farmacologia Aplicada À NutriçãoYam VictorNo ratings yet
- Divisão CelularDocument2 pagesDivisão CelularVictória DominguesNo ratings yet
- Interação Gênica E Herança Quantitativa: ComplementarDocument2 pagesInteração Gênica E Herança Quantitativa: ComplementarGabriela PiresNo ratings yet
- Cultivares 2013/2014Document41 pagesCultivares 2013/2014Filipe FariasNo ratings yet
- BacteriologiaDocument54 pagesBacteriologiaanonimoNo ratings yet
- Aula de Sinalização CelularDocument87 pagesAula de Sinalização CelularMarina Giusepette100% (1)
- HORUS Aula 11 BIOTECNOLOGIA - 113932Document2 pagesHORUS Aula 11 BIOTECNOLOGIA - 113932Kaua AmorimNo ratings yet
- Ariel Mariano SilberDocument15 pagesAriel Mariano SilberMijilovich KamarazovNo ratings yet
- Transdução de SinaisDocument6 pagesTransdução de SinaisthuanesgNo ratings yet
- Princípios Da FarmacodinâmicaDocument63 pagesPrincípios Da FarmacodinâmicaCelina PereiraNo ratings yet
- As Sequências-Sinal Direcionam As Proteínas para Os Compartimentos CorretosDocument4 pagesAs Sequências-Sinal Direcionam As Proteínas para Os Compartimentos CorretosRafael LuizNo ratings yet
- SP2 - A Dengue Ficou Por AquiDocument43 pagesSP2 - A Dengue Ficou Por AquiGeisa Maritsa WippelNo ratings yet
- Apresentação AntibioticosDocument54 pagesApresentação AntibioticosMatheus RodriguesNo ratings yet