You might also like
- Elementary Particles: The Commonwealth and International LibraryFrom EverandElementary Particles: The Commonwealth and International LibraryNo ratings yet
- A Very Brief History of The Study of Light: 1. Sir Isaac Newton 1672Document30 pagesA Very Brief History of The Study of Light: 1. Sir Isaac Newton 1672Niko DidicNo ratings yet
- Optics: International Series of Monographs in Natural PhilosophyFrom EverandOptics: International Series of Monographs in Natural PhilosophyRating: 3 out of 5 stars3/5 (1)
- CH-103-4th LectureDocument14 pagesCH-103-4th LectureDhruv DhirawaniNo ratings yet
- Macromolecular Microsymposium — 16: Main Lectures Presented at the Sixteenth Microsymposium on Macromolecules (Advances in Scattering Methods), Prague, 12 - 16 July 1976From EverandMacromolecular Microsymposium — 16: Main Lectures Presented at the Sixteenth Microsymposium on Macromolecules (Advances in Scattering Methods), Prague, 12 - 16 July 1976B. SedláčekNo ratings yet
- (Pre-Lab) Experiment 11 - Spectrophotometric AnalysisDocument23 pages(Pre-Lab) Experiment 11 - Spectrophotometric AnalysisPATRICIA ANJELIKA ANGELESNo ratings yet
- Essentials of Lasers: The Commonwealth and International Library: Selected Readings in PhysicsFrom EverandEssentials of Lasers: The Commonwealth and International Library: Selected Readings in PhysicsNo ratings yet
- Httpweb Iyte Edu TR Serifeyalcinlectureschem502L4 PDFDocument91 pagesHttpweb Iyte Edu TR Serifeyalcinlectureschem502L4 PDFAli AllamNo ratings yet
- Electron Paramagnetic Resonance in Modern Carbon-Based NanomaterialsFrom EverandElectron Paramagnetic Resonance in Modern Carbon-Based NanomaterialsNo ratings yet
- CYN-008 SpectrosDocument56 pagesCYN-008 SpectrosharshNo ratings yet
- Application of Molecular Absorption SpectrosDocument52 pagesApplication of Molecular Absorption SpectrosVeliana Teta100% (1)
- Uv VisibleDocument20 pagesUv VisibleahmedNo ratings yet
- E 6 Elec StructDocument50 pagesE 6 Elec StructJay-Rald LammataoNo ratings yet
- Raman ScatteringDocument65 pagesRaman ScatteringPragna ReddyNo ratings yet
- 1 PPSC UVVis and FTIR 1Document43 pages1 PPSC UVVis and FTIR 1Naeem AnwarNo ratings yet
- Rajesh Singh M. Pharm 1 Year: Made byDocument49 pagesRajesh Singh M. Pharm 1 Year: Made byNaman Sharma100% (1)
- SpectrosDocument58 pagesSpectrospaoloobiasNo ratings yet
- Absorption SpectrosDocument55 pagesAbsorption SpectrosfayvourajNo ratings yet
- Absorption SpectrosDocument55 pagesAbsorption SpectrosMahalakshmi SahasranamanNo ratings yet
- Excess Carriers in SemiconductorsDocument79 pagesExcess Carriers in SemiconductorsMayank SinghNo ratings yet
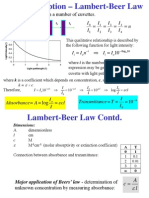
- Beers Law NotesDocument5 pagesBeers Law NotesJerome GironNo ratings yet
- Vibrational Spectroscopy: Modern Methods in Heterogeneous CatalysisDocument41 pagesVibrational Spectroscopy: Modern Methods in Heterogeneous CatalysisDevanesan KNo ratings yet
- Hotometry: Presenter: Dr. Anurag Yadav Moderator: Mr. Arun KumarDocument45 pagesHotometry: Presenter: Dr. Anurag Yadav Moderator: Mr. Arun KumarOsmmn KaleNo ratings yet
- Uv Visible SpectrosDocument48 pagesUv Visible SpectrosDhanvanth100% (7)
- Spectroscopy L1Document74 pagesSpectroscopy L1Yashvi ChauhanNo ratings yet
- 21 9 6 6. Chapter 13. Ultraviolet Visible Spectrophotometry 11.29Document16 pages21 9 6 6. Chapter 13. Ultraviolet Visible Spectrophotometry 11.29nekenih413No ratings yet
- Ltraviolet Isible Pectroscopy: Conversion To AbsorbanceDocument9 pagesLtraviolet Isible Pectroscopy: Conversion To AbsorbanceCarolNachNo ratings yet
- The Interaction of Light and MatterDocument19 pagesThe Interaction of Light and MatterIbrahim MoradNo ratings yet
- Spectroscopy UV Via ND IR f1 PDFDocument91 pagesSpectroscopy UV Via ND IR f1 PDFJanakiramNo ratings yet
- 5 Edition. Philippines: Elsevier)Document3 pages5 Edition. Philippines: Elsevier)Daren SantosNo ratings yet
- Atomic and Molecular Spectroscopy-2 PDFDocument60 pagesAtomic and Molecular Spectroscopy-2 PDFHaseeb HaiderNo ratings yet
- Uv Visible SpectrosDocument14 pagesUv Visible SpectrosDevanshi JadaunNo ratings yet
- Uv Visible SpectrosDocument14 pagesUv Visible SpectrosDevanshi JadaunNo ratings yet
- Part 3 Using Light For DetectionDocument48 pagesPart 3 Using Light For DetectionJosé ClementeNo ratings yet
- Lect2-Light ReactionDocument62 pagesLect2-Light ReactionGilang Satya ArdhanaNo ratings yet
- Instrumental Lecture 2Document114 pagesInstrumental Lecture 2Serhat Burak KarasakalNo ratings yet
- UV-visible Molecular Absorption SpectrosDocument33 pagesUV-visible Molecular Absorption SpectrosaargovindNo ratings yet
- Spectroscopic MethodsDocument76 pagesSpectroscopic MethodsVu SangNo ratings yet
- Chap1 UV-VIS LectureNoteDocument21 pagesChap1 UV-VIS LectureNoteAby JatNo ratings yet
- 1.UV Spectro - IntroductionDocument49 pages1.UV Spectro - IntroductionRitika GuptaNo ratings yet
- IR and UV SpectroDocument69 pagesIR and UV SpectroSk KumarNo ratings yet
- Department of Chemistry: Michael J. HynesDocument55 pagesDepartment of Chemistry: Michael J. Hynesfahim khattakNo ratings yet
- Theory of Ultraviolet SpectrosDocument49 pagesTheory of Ultraviolet Spectrosanusha jrNo ratings yet
- Uv / V S: Isible PectrosDocument57 pagesUv / V S: Isible PectrosTahreem FatimaNo ratings yet
- An Introduction To Spectroscopy & Molecular Spectroscopy: Analytical Chemistry LectureDocument39 pagesAn Introduction To Spectroscopy & Molecular Spectroscopy: Analytical Chemistry Lecture陳庠廷No ratings yet
- Basic Spectroscopy LectureDocument134 pagesBasic Spectroscopy LectureMa Rian CombineNo ratings yet
- Doppler-Free Saturation Spectroscopy: Thomas Rieger and Thomas VolzDocument23 pagesDoppler-Free Saturation Spectroscopy: Thomas Rieger and Thomas Volzhoangloc NgoNo ratings yet
- 12 04 2006 Biophysics OptSpec 3hrDocument54 pages12 04 2006 Biophysics OptSpec 3hrapi-3696530No ratings yet
- Dept. of App. Chem. DTUDocument63 pagesDept. of App. Chem. DTUB1605 SAKSHAM MISHRANo ratings yet
- RUBY ExpDocument7 pagesRUBY ExpWan AizuddinNo ratings yet
- Colorimetry: Absorbance Is Directly Proportional To Concentration of FeDocument38 pagesColorimetry: Absorbance Is Directly Proportional To Concentration of Fedhekle_dNo ratings yet
- JSL-Lect 5 - UV-Vis-30-10-21Document25 pagesJSL-Lect 5 - UV-Vis-30-10-21Divyansh SharmaNo ratings yet
- Unit - IDocument19 pagesUnit - IManikandan KNo ratings yet
- Transisi ElektronikDocument122 pagesTransisi ElektronikChacha Chuka Smashclama'xNo ratings yet
- Lecture 7Document30 pagesLecture 7Raj KumarNo ratings yet
- Atomic Spectroscopy-CoordinatorDocument35 pagesAtomic Spectroscopy-Coordinatordsofatbe23No ratings yet
- Karakterisasi - Material - UV-Visible Spectroscopy - 240206 - 114342Document22 pagesKarakterisasi - Material - UV-Visible Spectroscopy - 240206 - 114342friska.ziliwu2590No ratings yet
- Uv-VISIBLE SPECTROSDocument41 pagesUv-VISIBLE SPECTROSVansh YadavNo ratings yet
- An Introduction To Infrared and UV-Visible SpectrosDocument45 pagesAn Introduction To Infrared and UV-Visible SpectrosArvandz_tea100% (1)
- UV-VIS Absorption Spectroscopy (Electronic Spectroscopy) : The Earliest Method of Molecular Spectroscopy!Document62 pagesUV-VIS Absorption Spectroscopy (Electronic Spectroscopy) : The Earliest Method of Molecular Spectroscopy!Niko DidicNo ratings yet
- Direct-Current Glow Discharges in Atmospheric Pressure Air PlasmasDocument10 pagesDirect-Current Glow Discharges in Atmospheric Pressure Air Plasmaseze_firmatenseNo ratings yet
- Histopathological Image Analysis: A ReviewDocument25 pagesHistopathological Image Analysis: A ReviewwebsternhidzaNo ratings yet
- Low Energy Electron Diffraction: & 2005, Elsevier Ltd. All Rights ReservedDocument11 pagesLow Energy Electron Diffraction: & 2005, Elsevier Ltd. All Rights ReservedSohidul MondalNo ratings yet
- Electron Spin Resonance SpectrosDocument29 pagesElectron Spin Resonance Spectroslalithawill100% (1)
- BYCHKOV - The Atmosphere and IonosphereDocument387 pagesBYCHKOV - The Atmosphere and IonosphereNestor HidalgoNo ratings yet
- Deconvolution Technique Application To Spectral Contour AnalysisDocument12 pagesDeconvolution Technique Application To Spectral Contour AnalysisYassine ZidNo ratings yet
- Applications of Uv in ClinicalDocument13 pagesApplications of Uv in ClinicalAnonymous Z7myJpGfRNo ratings yet
- Raman Spectrometric PAT Models Successful Transfer From Minibioreactors To Larger-Scale, Stirred-Tank BioreactorsDocument6 pagesRaman Spectrometric PAT Models Successful Transfer From Minibioreactors To Larger-Scale, Stirred-Tank BioreactorsKhaja Riazuddin Nawaz MohammedNo ratings yet
- InSightX3Plus DatasheetDocument4 pagesInSightX3Plus DatasheetĐình ThắngNo ratings yet
- Phosphorescence Excitation Spectrum of Benzophenone at Liq.N TemperatureDocument5 pagesPhosphorescence Excitation Spectrum of Benzophenone at Liq.N TemperatureNisar Ali Mphil-Chem ANo ratings yet
- CHEM340 Tut AAS With AnswersDocument4 pagesCHEM340 Tut AAS With AnswersAlex Tan100% (3)
- Spettro Infrarosso Del KCLO3Document6 pagesSpettro Infrarosso Del KCLO3AlessandroNo ratings yet
- MRIDocument59 pagesMRIUicheul YoonNo ratings yet
- Use of The Spectrophotometer-2Document7 pagesUse of The Spectrophotometer-2Tori Andika BukitNo ratings yet
- Spectrophotometric Determination of Iron Using 1,10-PhenanthrolineDocument9 pagesSpectrophotometric Determination of Iron Using 1,10-PhenanthrolineVeronique PicardNo ratings yet
- Hsslive - Plus Two Chapter 12-2024Document7 pagesHsslive - Plus Two Chapter 12-202416739No ratings yet
- FTIR AssignmentDocument4 pagesFTIR AssignmentAmirul Assyraf NoorNo ratings yet
- The Reason For The Excess Chelate in The OmniscanDocument4 pagesThe Reason For The Excess Chelate in The OmniscangasfgdNo ratings yet
- Biomimetically-Mineralized Composite Coatings On Titanium Functionalized With Gelatin Methacrylate HydrogelsDocument8 pagesBiomimetically-Mineralized Composite Coatings On Titanium Functionalized With Gelatin Methacrylate Hydrogelsabdul basitNo ratings yet
- SOx in Ambient AirDocument10 pagesSOx in Ambient AirKushal SharmaNo ratings yet
- Analytical Instruments Question BankDocument18 pagesAnalytical Instruments Question BankElaineNo ratings yet
- Pha052 TG 7Document7 pagesPha052 TG 7Alcea InguilloNo ratings yet
- ChT-Additional-Questions-from-Sanfoundry-keyclean 3Document25 pagesChT-Additional-Questions-from-Sanfoundry-keyclean 3Reinalyn AnyayahanNo ratings yet
- TMP 610 CDocument6 pagesTMP 610 CFrontiersNo ratings yet
- En Physics Laboratory Experiments Tess ExpertDocument324 pagesEn Physics Laboratory Experiments Tess Expertpolyyking100% (1)
- Cerchiaro 2013Document14 pagesCerchiaro 2013Osvaldo RomanNo ratings yet
- TiO2 Synthesis Using GlycineDocument5 pagesTiO2 Synthesis Using GlycineRanjit KumarNo ratings yet
- Spectrochimica Acta Part A: Molecular and Biomolecular SpectrosDocument11 pagesSpectrochimica Acta Part A: Molecular and Biomolecular SpectrosPavan KumarNo ratings yet
- Vibrational Spectroscopy of Biological and Polymeric MaterialsDocument447 pagesVibrational Spectroscopy of Biological and Polymeric MaterialsZurd_oNo ratings yet
- Precila C.F. Ip Et Al - Optical-Optical Double-Resonance Spectroscopy of BaF: The E 2-Sigma + and F 2-Pi StatesDocument9 pagesPrecila C.F. Ip Et Al - Optical-Optical Double-Resonance Spectroscopy of BaF: The E 2-Sigma + and F 2-Pi StatesUasnsdaNo ratings yet
- ICH Quality Guidelines: An Implementation GuideFrom EverandICH Quality Guidelines: An Implementation GuideAndrew TeasdaleNo ratings yet
- The Regenerative Grower's Guide to Garden Amendments: Using Locally Sourced Materials to Make Mineral and Biological Extracts and FermentsFrom EverandThe Regenerative Grower's Guide to Garden Amendments: Using Locally Sourced Materials to Make Mineral and Biological Extracts and FermentsRating: 5 out of 5 stars5/5 (3)
- The Nature of Drugs Vol. 1: History, Pharmacology, and Social ImpactFrom EverandThe Nature of Drugs Vol. 1: History, Pharmacology, and Social ImpactRating: 5 out of 5 stars5/5 (5)
- Periodic Tales: A Cultural History of the Elements, from Arsenic to ZincFrom EverandPeriodic Tales: A Cultural History of the Elements, from Arsenic to ZincRating: 3.5 out of 5 stars3.5/5 (137)
- Handbook of Formulating Dermal Applications: A Definitive Practical GuideFrom EverandHandbook of Formulating Dermal Applications: A Definitive Practical GuideNo ratings yet
- Chemistry for Breakfast: The Amazing Science of Everyday LifeFrom EverandChemistry for Breakfast: The Amazing Science of Everyday LifeRating: 4.5 out of 5 stars4.5/5 (14)
- The Nature of Drugs Vol. 1: History, Pharmacology, and Social ImpactFrom EverandThe Nature of Drugs Vol. 1: History, Pharmacology, and Social ImpactRating: 5 out of 5 stars5/5 (1)
- It's Elemental: The Hidden Chemistry in EverythingFrom EverandIt's Elemental: The Hidden Chemistry in EverythingRating: 4 out of 5 stars4/5 (10)
- Chemistry: a QuickStudy Laminated Reference GuideFrom EverandChemistry: a QuickStudy Laminated Reference GuideRating: 5 out of 5 stars5/5 (1)
- AP Chemistry Flashcards, Fourth Edition: Up-to-Date Review and PracticeFrom EverandAP Chemistry Flashcards, Fourth Edition: Up-to-Date Review and PracticeNo ratings yet
- Guidelines for Defining Process Safety Competency RequirementsFrom EverandGuidelines for Defining Process Safety Competency RequirementsRating: 3 out of 5 stars3/5 (1)
- AP® Chemistry Crash Course, For the 2020 Exam, Book + Online: Get a Higher Score in Less TimeFrom EverandAP® Chemistry Crash Course, For the 2020 Exam, Book + Online: Get a Higher Score in Less TimeRating: 5 out of 5 stars5/5 (1)
- The Production of Volatile Oils and Perfumery Plants in the United StatesFrom EverandThe Production of Volatile Oils and Perfumery Plants in the United StatesNo ratings yet
- Monkeys, Myths, and Molecules: Separating Fact from Fiction, and the Science of Everyday LifeFrom EverandMonkeys, Myths, and Molecules: Separating Fact from Fiction, and the Science of Everyday LifeRating: 4 out of 5 stars4/5 (1)
- Chemistry for Breakfast: The Amazing Science of Everyday LifeFrom EverandChemistry for Breakfast: The Amazing Science of Everyday LifeRating: 4.5 out of 5 stars4.5/5 (90)
- Taste: Surprising Stories and Science About Why Food Tastes GoodFrom EverandTaste: Surprising Stories and Science About Why Food Tastes GoodRating: 3 out of 5 stars3/5 (20)
- Essential Chemistry for Formulators of Semisolid and Liquid DosagesFrom EverandEssential Chemistry for Formulators of Semisolid and Liquid DosagesRating: 5 out of 5 stars5/5 (2)