You might also like
- Lithospheric DiscontinuitiesFrom EverandLithospheric DiscontinuitiesHuaiyu YuanNo ratings yet
- tmp693 TMPDocument17 pagestmp693 TMPFrontiersNo ratings yet
- Jurnal Acuan CO2 Flux Pak Aan (Coret)Document35 pagesJurnal Acuan CO2 Flux Pak Aan (Coret)Echa Priyaning AryunishaNo ratings yet
- Geiger 2013Document8 pagesGeiger 2013Rafi Uddin RaiyanNo ratings yet
- Climate Pacing of Millennial Sea-Level Change Variability in The Central and Western MediterraneanDocument9 pagesClimate Pacing of Millennial Sea-Level Change Variability in The Central and Western Mediterraneanat taNo ratings yet
- Water 05 01303Document23 pagesWater 05 01303Juan David OrtizNo ratings yet
- McKinley 2016 Carbon SinkDocument12 pagesMcKinley 2016 Carbon SinkgopikasuresNo ratings yet
- Modelling Long-Term Changes in Stream Chemistry for an Acidic Welsh MoorlandDocument14 pagesModelling Long-Term Changes in Stream Chemistry for an Acidic Welsh MoorlandCAMILONo ratings yet
- 2009 - Decadal Increase of Oceanic Carbon Dioxide in Southern Indian Ocean Surface Waters (1991-2007)Document13 pages2009 - Decadal Increase of Oceanic Carbon Dioxide in Southern Indian Ocean Surface Waters (1991-2007)donny sophandiNo ratings yet
- Remote Sensing of Sea Surface Salinity: An Overview With Case StudiesDocument10 pagesRemote Sensing of Sea Surface Salinity: An Overview With Case StudiesZulkifli i. TuaraNo ratings yet
- Churchill - The Dynamics of Weather-Band Sea Level Variations in The Red SeaDocument7 pagesChurchill - The Dynamics of Weather-Band Sea Level Variations in The Red SeaAtef AlshehriNo ratings yet
- Carter 2016 Ocean AcidificationDocument18 pagesCarter 2016 Ocean AcidificationgopikasuresNo ratings yet
- Presentation MartinsDocument29 pagesPresentation MartinsAlexNo ratings yet
- Quantifying Groundwater Discharge To Cockburn River, Southeastern Australia, Using Dissolved Gas Tracers222rn and SF6Document12 pagesQuantifying Groundwater Discharge To Cockburn River, Southeastern Australia, Using Dissolved Gas Tracers222rn and SF6Eder Queiroz BarbosaNo ratings yet
- Ocean Acidification ThesisDocument12 pagesOcean Acidification Thesisevkrjniig100% (1)
- Relationship Between Salinity and Flow On Dissolved Oxygen Case StudyDocument12 pagesRelationship Between Salinity and Flow On Dissolved Oxygen Case StudyevanNo ratings yet
- 5.4 Marine Biodiversity and Ecosystems: An Indicator-Based AssessmentDocument35 pages5.4 Marine Biodiversity and Ecosystems: An Indicator-Based AssessmentVesnaBarbuliNo ratings yet
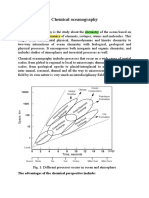
- CHEMICAL OCEANOGRAPHYDocument8 pagesCHEMICAL OCEANOGRAPHYHalima akterNo ratings yet
- Vertical Variability of Near-Surface Salinity in The Tropics: Consequences For L-Band Radiometer Calibration and ValidationDocument18 pagesVertical Variability of Near-Surface Salinity in The Tropics: Consequences For L-Band Radiometer Calibration and ValidationOceans HackathonNo ratings yet
- Keraghel 2020Document16 pagesKeraghel 2020dj SidNo ratings yet
- Department of Geophysical Sciences, University of Chicago, IllinoisDocument26 pagesDepartment of Geophysical Sciences, University of Chicago, IllinoisArne WinguthNo ratings yet
- Limnology Oceanography: MethodsDocument21 pagesLimnology Oceanography: MethodsStefano PontarelliNo ratings yet
- Global Biogeochemical Cycles - 2022 - Davila - How Is The Ocean Anthropogenic Carbon Reservoir FilledDocument16 pagesGlobal Biogeochemical Cycles - 2022 - Davila - How Is The Ocean Anthropogenic Carbon Reservoir Filled2646501812No ratings yet
- CooperetalJGR 05Document14 pagesCooperetalJGR 052003600rrNo ratings yet
- Heinze 2018 Ocean AcidificationDocument19 pagesHeinze 2018 Ocean AcidificationgopikasuresNo ratings yet
- Researc Project Proposal On Sediment TransportsDocument7 pagesResearc Project Proposal On Sediment TransportsZafirahNo ratings yet
- Geochemistry of West Siberian Streams and Their Potential Response To Permafrost DegradationDocument15 pagesGeochemistry of West Siberian Streams and Their Potential Response To Permafrost DegradationEkaStaVTVNo ratings yet
- Aa-Am: Aabw Aacw Aaiw Aasw Aate Abiki ABF ABL ABP Absolute HumidityDocument23 pagesAa-Am: Aabw Aacw Aaiw Aasw Aate Abiki ABF ABL ABP Absolute HumidityRicardo Saldaña PausicNo ratings yet
- 1 s2.0 S0016703711005047 MainDocument16 pages1 s2.0 S0016703711005047 MainManoelsonNo ratings yet
- Continental Shelf Research: Sophie L. Ward, James D. Scourse, Yusuke Yokoyama, Simon P. NeillDocument10 pagesContinental Shelf Research: Sophie L. Ward, James D. Scourse, Yusuke Yokoyama, Simon P. Neillfaris nauvalNo ratings yet
- Pandolfi Et Al 2011 Science 2011Document6 pagesPandolfi Et Al 2011 Science 2011Muliari AyiNo ratings yet
- Simulation of Groundwater-Seawater Interaction in The Coastal Surficial Aquifer in Bohai Bay, Tianjin, China (2016)Document11 pagesSimulation of Groundwater-Seawater Interaction in The Coastal Surficial Aquifer in Bohai Bay, Tianjin, China (2016)Anonymous 291PWogNo ratings yet
- Articulo AnaliticaDocument16 pagesArticulo AnaliticaPaula VelandiaNo ratings yet
- Shrinkingof Fishes Exacerbates Impacts of Global Ocean Changes On Marine Ecosystems Sep2012 CheungDocument5 pagesShrinkingof Fishes Exacerbates Impacts of Global Ocean Changes On Marine Ecosystems Sep2012 CheungJorge GrandezNo ratings yet
- Chemical and Physical Transformations of Mercury in The Ocean A ReviewDocument17 pagesChemical and Physical Transformations of Mercury in The Ocean A ReviewyanyuliNo ratings yet
- Modelling The Impacts of Climate Change On An Eroding Coast Over The 21st CenturyDocument34 pagesModelling The Impacts of Climate Change On An Eroding Coast Over The 21st CenturyTyndall Centre for Climate Change ResearchNo ratings yet
- PDF Original 3Document12 pagesPDF Original 3MARCO ENCISO HUAMANNo ratings yet
- Lutz - Et - al-2007-JGR POC Flux PDFDocument26 pagesLutz - Et - al-2007-JGR POC Flux PDFBella MorteNo ratings yet
- Geosciences 10 00506Document7 pagesGeosciences 10 00506Cleber Dos SantosNo ratings yet
- Comparison of Light Hydrocarbon Microseepage MechanismsDocument12 pagesComparison of Light Hydrocarbon Microseepage Mechanismsqiangeng007No ratings yet
- Global Biogeochemical Cycles - 2018 - Wolf - Oxygen Saturation Surrounding Deep Water Formation Events in The Labrador SeaDocument19 pagesGlobal Biogeochemical Cycles - 2018 - Wolf - Oxygen Saturation Surrounding Deep Water Formation Events in The Labrador Seanovia putri andikaNo ratings yet
- Glossary of Physical Oceanography and Related Disciplines - Steven K. BaumDocument517 pagesGlossary of Physical Oceanography and Related Disciplines - Steven K. BaumLuis Gerardo Escandon Alcazar100% (1)
- Coastal and Sediment TransportDocument5 pagesCoastal and Sediment TransportSianly ImapulyNo ratings yet
- Dissolved Organic Matter Characterisation and Temporal Trends in Terra Nova Bay (Ross Sea, Antarctica)Document10 pagesDissolved Organic Matter Characterisation and Temporal Trends in Terra Nova Bay (Ross Sea, Antarctica)amensetNo ratings yet
- EAS 4300: Introduction To Oceanography, FALL 2009: InstructorsDocument23 pagesEAS 4300: Introduction To Oceanography, FALL 2009: InstructorsPankaj MunjalNo ratings yet
- Descarga Del MagdalenaDocument17 pagesDescarga Del MagdalenaEdid MedinaNo ratings yet
- 7 1 HollandDocument2 pages7 1 HollandnvanthaoNo ratings yet
- Gral 22Document12 pagesGral 22claudioNo ratings yet
- Zhang Church-2012Document8 pagesZhang Church-2012toreba3568No ratings yet
- Filtering Methods in Tidal-Affected Groundwater Head Measurements: Application of Harmonic Analysis and Continuous Wavelet TransformDocument38 pagesFiltering Methods in Tidal-Affected Groundwater Head Measurements: Application of Harmonic Analysis and Continuous Wavelet TransformMuhamad IrvanNo ratings yet
- An Introduction To Causes and Consequences of Cretaceous Sea-Level Changes (IGCP 609)Document8 pagesAn Introduction To Causes and Consequences of Cretaceous Sea-Level Changes (IGCP 609)Soltani AkRêmNo ratings yet
- Diccionary For Oceanography.Document539 pagesDiccionary For Oceanography.RaquelSanchezNo ratings yet
- 8 PETM Abrupt-EventsDocument4 pages8 PETM Abrupt-EventsBrandel CoolenNo ratings yet
- Salinity Paper JCR-2Document10 pagesSalinity Paper JCR-2kokakolakkiyaNo ratings yet
- tmpE8A4 TMPDocument11 pagestmpE8A4 TMPFrontiersNo ratings yet
- Seawater Carbon and Strontium Isotope Variations Through The Late Ediacaran To Late Cambrian in The Tarim BasinDocument59 pagesSeawater Carbon and Strontium Isotope Variations Through The Late Ediacaran To Late Cambrian in The Tarim BasinYinggang ZhangNo ratings yet
- Climate and vegetation reconstructions from European sedimentsDocument1 pageClimate and vegetation reconstructions from European sedimentsabdounouNo ratings yet
- Limnology Ocean Methods - 2009 - Berg - Eddy correlation measurements of oxygen uptake in deep ocean sedimentsDocument9 pagesLimnology Ocean Methods - 2009 - Berg - Eddy correlation measurements of oxygen uptake in deep ocean sedimentsDiego Alejandro Pulgarin MontoyaNo ratings yet
- Bitzin 2005Document1 pageBitzin 2005Nicholas Matthew WelshNo ratings yet
- Lecture 01Document11 pagesLecture 01Hilmi HasaniNo ratings yet
- Exclusive - Tiger Temple Accused of Supplying Black MarketDocument16 pagesExclusive - Tiger Temple Accused of Supplying Black Marketsharon_guynupNo ratings yet
- Journalists Need To Do More To Cover Wildlife and Environmental CrimeDocument6 pagesJournalists Need To Do More To Cover Wildlife and Environmental Crimesharon_guynupNo ratings yet
- Pangolins On The Brink As Africa-China Trafficking Persists UnabatedDocument30 pagesPangolins On The Brink As Africa-China Trafficking Persists Unabatedsharon_guynupNo ratings yet
- Tigers in The TankDocument6 pagesTigers in The Tanksharon_guynupNo ratings yet
- Listening Comprehension (30 Minutes) : Section ADocument12 pagesListening Comprehension (30 Minutes) : Section A威尔斯查德No ratings yet
- Landforms With PicsDocument5 pagesLandforms With PicsmllorenteNo ratings yet
- Essay On ConductionDocument4 pagesEssay On Conductionnzila43No ratings yet
- LE Science 5 MELC 9Document6 pagesLE Science 5 MELC 9GERWIN MARIANONo ratings yet
- Reading Passage 1: IELTS Recent Actual Test With Answers Volume 6Document15 pagesReading Passage 1: IELTS Recent Actual Test With Answers Volume 6Hajer DiefNo ratings yet
- Appeal No.100 of 2015 (SZ) - National Green Tribunal - JudgmentDocument34 pagesAppeal No.100 of 2015 (SZ) - National Green Tribunal - Judgmentkayalontheweb100% (1)
- Act No 6 Year 1996 On Indonesian WatersDocument11 pagesAct No 6 Year 1996 On Indonesian Watersindra alverdianNo ratings yet
- Appendix A3: Phil-IRI Group Screening Tests in EnglishDocument14 pagesAppendix A3: Phil-IRI Group Screening Tests in EnglishDahina Cayabas100% (1)
- Instruction Manual Echo Sounder JFE-680 PDFDocument62 pagesInstruction Manual Echo Sounder JFE-680 PDFHenrysusanto Susanto0% (1)
- Jurnal JmasDocument26 pagesJurnal Jmasyama wiratamaNo ratings yet
- Ecosystems 2Document25 pagesEcosystems 2HUDA FATHIMA 2031826No ratings yet
- Harbour & Its Classification Page - 1Document6 pagesHarbour & Its Classification Page - 1deepu1009No ratings yet
- HVDC ELECTRODE DESIGN - Cigre (675 PDFDocument150 pagesHVDC ELECTRODE DESIGN - Cigre (675 PDFAsif Ali100% (1)
- The International and Domestic Law Basis For The Shared Conservation, Management and Use of Sea Turtles in Nicaragua, Costa Rica and PanamaDocument156 pagesThe International and Domestic Law Basis For The Shared Conservation, Management and Use of Sea Turtles in Nicaragua, Costa Rica and PanamaPrograma Regional para el Manejo de Recursos Acuáticos y Alternativas EconómicasNo ratings yet
- The Environment Vocabulary Practice - 80587Document2 pagesThe Environment Vocabulary Practice - 80587Elena ContrasNo ratings yet
- SDG14 Life Below Water - What Are The Type of Bodies of WaterDocument16 pagesSDG14 Life Below Water - What Are The Type of Bodies of WaterEdbonessasangNo ratings yet
- Munyua, M - Determining The Magnitude of Wind Loads On Structures in Kenya According To The Eurocodes 2021-01-12Document8 pagesMunyua, M - Determining The Magnitude of Wind Loads On Structures in Kenya According To The Eurocodes 2021-01-12opulitheNo ratings yet
- Land and Water Forms of The WorldDocument12 pagesLand and Water Forms of The WorldXenonexNo ratings yet
- Assessment Capt. VinoDocument86 pagesAssessment Capt. VinoClara suci atiaraNo ratings yet
- Masud Perbas Assignment On Bangladesh Tourism Saitn MartinDocument10 pagesMasud Perbas Assignment On Bangladesh Tourism Saitn Martinimrul khanNo ratings yet
- Concentration Factor of Metals in Zooplankton and Their Seasonality in Kalpakkam Coast, Southwest Bay of BengalDocument12 pagesConcentration Factor of Metals in Zooplankton and Their Seasonality in Kalpakkam Coast, Southwest Bay of BengalSatya PanigrahiNo ratings yet
- TheWorldontheTurtlesBack 1Document8 pagesTheWorldontheTurtlesBack 1Kristen GoddardNo ratings yet
- Đề Chính ThứcDocument15 pagesĐề Chính ThứcBach Hua HuaNo ratings yet
- The Biblical Flood and The Ice EpochDocument2 pagesThe Biblical Flood and The Ice EpochCraig Peters50% (2)
- Vocabulary Related To Unit4Tthe Environment.Document2 pagesVocabulary Related To Unit4Tthe Environment.Omar Belazri0% (1)
- Restate Key Words - Answer All Parts: All Questions Have Specific KEYWORDS. Some Questions Have Multiple PartsDocument3 pagesRestate Key Words - Answer All Parts: All Questions Have Specific KEYWORDS. Some Questions Have Multiple PartsFelicia DavisNo ratings yet
- Thesis Statement About Flash FloodDocument8 pagesThesis Statement About Flash Flooddwsdzrcq100% (2)
- Harbors Design ManualDocument132 pagesHarbors Design Manualvasu7900100% (1)
- Coastal Assignment EDITED 4Document24 pagesCoastal Assignment EDITED 4mohamadfaiz0% (1)
- Bahasa Inggris 9Document8 pagesBahasa Inggris 9muslimNo ratings yet