You might also like
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryFrom EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryRating: 3.5 out of 5 stars3.5/5 (231)
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)From EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Rating: 4.5 out of 5 stars4.5/5 (119)
- Never Split the Difference: Negotiating As If Your Life Depended On ItFrom EverandNever Split the Difference: Negotiating As If Your Life Depended On ItRating: 4.5 out of 5 stars4.5/5 (838)
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaFrom EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaRating: 4.5 out of 5 stars4.5/5 (265)
- The Little Book of Hygge: Danish Secrets to Happy LivingFrom EverandThe Little Book of Hygge: Danish Secrets to Happy LivingRating: 3.5 out of 5 stars3.5/5 (399)
- Grit: The Power of Passion and PerseveranceFrom EverandGrit: The Power of Passion and PerseveranceRating: 4 out of 5 stars4/5 (587)
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyFrom EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyRating: 3.5 out of 5 stars3.5/5 (2219)
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeFrom EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeRating: 4 out of 5 stars4/5 (5794)
- Team of Rivals: The Political Genius of Abraham LincolnFrom EverandTeam of Rivals: The Political Genius of Abraham LincolnRating: 4.5 out of 5 stars4.5/5 (234)
- Shoe Dog: A Memoir by the Creator of NikeFrom EverandShoe Dog: A Memoir by the Creator of NikeRating: 4.5 out of 5 stars4.5/5 (537)
- The Emperor of All Maladies: A Biography of CancerFrom EverandThe Emperor of All Maladies: A Biography of CancerRating: 4.5 out of 5 stars4.5/5 (271)
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreFrom EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreRating: 4 out of 5 stars4/5 (1090)
- Her Body and Other Parties: StoriesFrom EverandHer Body and Other Parties: StoriesRating: 4 out of 5 stars4/5 (821)
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersFrom EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersRating: 4.5 out of 5 stars4.5/5 (344)
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceFrom EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceRating: 4 out of 5 stars4/5 (890)
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureFrom EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureRating: 4.5 out of 5 stars4.5/5 (474)
- The Unwinding: An Inner History of the New AmericaFrom EverandThe Unwinding: An Inner History of the New AmericaRating: 4 out of 5 stars4/5 (45)
- The Yellow House: A Memoir (2019 National Book Award Winner)From EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Rating: 4 out of 5 stars4/5 (98)
- On Fire: The (Burning) Case for a Green New DealFrom EverandOn Fire: The (Burning) Case for a Green New DealRating: 4 out of 5 stars4/5 (73)
- El Bill PDFDocument2 pagesEl Bill PDFvinodNo ratings yet
- SCAT Chart - Systematic Cause Analysis Technique - SCAT ChartDocument6 pagesSCAT Chart - Systematic Cause Analysis Technique - SCAT ChartSalman Alfarisi100% (1)
- Hospital & Clinical Pharmacy Q&ADocument22 pagesHospital & Clinical Pharmacy Q&AKrishan KumarNo ratings yet
- 5 S Principles ExplainedDocument30 pages5 S Principles Explainedamaresh nkNo ratings yet
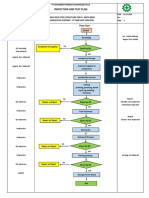
- Inspection and Test Plan: Flow Chart Start IncomingDocument1 pageInspection and Test Plan: Flow Chart Start IncomingSinden AyuNo ratings yet
- Electrical Interview Questions & Answers - Hydro Power PlantDocument2 pagesElectrical Interview Questions & Answers - Hydro Power PlantLaxman Naidu NNo ratings yet
- Presentation On Data Integrity in PharmaDocument80 pagesPresentation On Data Integrity in Pharmaskvemula67% (3)
- DNV Rules For Electrical Instal at IonsDocument80 pagesDNV Rules For Electrical Instal at Ionsnzjohn100% (3)
- RESEARCH PROPOSAL-Final AfraaaazzzzzzzzzDocument13 pagesRESEARCH PROPOSAL-Final AfraaaazzzzzzzzzRizwan Abdul Maalik50% (2)
- Cswip Visual Welding Inspector 3.0 BrochureDocument1 pageCswip Visual Welding Inspector 3.0 BrochureNasfauzan100% (2)
- Eltra Cs 530Document122 pagesEltra Cs 530ahalonsoNo ratings yet
- Steroids ActivityDocument1 pageSteroids Activityfaqed ilzakiraNo ratings yet
- T1D Report September 2023Document212 pagesT1D Report September 2023Andrei BombardieruNo ratings yet
- Husqvarna 340/345/350 Operator's ManualDocument47 pagesHusqvarna 340/345/350 Operator's ManualArtur MartinsNo ratings yet
- Human Diseases A Systemic Approach 8th Edition-Páginas-15-26Document12 pagesHuman Diseases A Systemic Approach 8th Edition-Páginas-15-26Karime LopezNo ratings yet
- Nursing Care of ElderlyDocument26 pagesNursing Care of ElderlyIndra KumarNo ratings yet
- 01 01Document232 pages01 01Muhammad Al-MshariNo ratings yet
- Adolescent and Sexual HealthDocument36 pagesAdolescent and Sexual Healthqwerty123No ratings yet
- Report Experiment 5 STK1211Document9 pagesReport Experiment 5 STK1211NurAkila Mohd YasirNo ratings yet
- Fiitjee JEE Adv p1 Phase II SolDocument10 pagesFiitjee JEE Adv p1 Phase II SolPadamNo ratings yet
- Cd6352 Lee Swee LeongDocument24 pagesCd6352 Lee Swee LeongFrea Marie SarabiaNo ratings yet
- 21 - Effective Pages: Beechcraft CorporationDocument166 pages21 - Effective Pages: Beechcraft CorporationCristian PugaNo ratings yet
- Ethnobotany Manual 14th September 2016Document54 pagesEthnobotany Manual 14th September 2016Rahul0% (1)
- Jee Main Sample Paper 5Document19 pagesJee Main Sample Paper 5DavidNo ratings yet
- Khatr Khola ISP District RatesDocument56 pagesKhatr Khola ISP District RatesCivil EngineeringNo ratings yet
- Mole Concept: Chemfile Mini-Guide To Problem SolvingDocument18 pagesMole Concept: Chemfile Mini-Guide To Problem SolvingNaren ParasharNo ratings yet
- Coal Workers' Pneumoconiosis (Black Lung Disease) Treatment & Management - Approach Considerations, Medical Care, Surgical CareDocument2 pagesCoal Workers' Pneumoconiosis (Black Lung Disease) Treatment & Management - Approach Considerations, Medical Care, Surgical CareامينNo ratings yet
- Temporomandibular Joint SyndromeDocument11 pagesTemporomandibular Joint SyndromeRahma RahmaNo ratings yet
- Requirement & Other Requirement: 2.311 Procedure For Accessing Applicable LegalDocument2 pagesRequirement & Other Requirement: 2.311 Procedure For Accessing Applicable Legalkirandevi1981No ratings yet
- RCF's Market Analysis of Dealer NetworkDocument70 pagesRCF's Market Analysis of Dealer NetworkAshok KushwahaNo ratings yet