You might also like
- Emergency Psychiatry Other Than Suicide: Dr. Pooja Singh, MD Assistant ProfessorDocument45 pagesEmergency Psychiatry Other Than Suicide: Dr. Pooja Singh, MD Assistant Professorpooja singhNo ratings yet
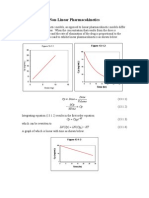
- Non Linear PharmacokineticsDocument94 pagesNon Linear PharmacokineticsJaspreet Guraya100% (1)
- Bioavailability and BioequivalenceDocument82 pagesBioavailability and Bioequivalenceكسلان اكتب اسمي100% (1)
- Introduction To BiopharmaceuticsDocument106 pagesIntroduction To BiopharmaceuticsHely Patel100% (1)
- Mercury Lead Arsenic Cadmium ToxicityDocument171 pagesMercury Lead Arsenic Cadmium ToxicitySunilNo ratings yet
- Drugs Acting On Digestive System of AnimalsDocument11 pagesDrugs Acting On Digestive System of AnimalsSunil100% (3)
- AntiConvulsants Drugs in Brief PDFDocument28 pagesAntiConvulsants Drugs in Brief PDFSunilNo ratings yet
- Pharmacokinetic ModelsDocument36 pagesPharmacokinetic ModelsNeeraj Kumar100% (1)
- BIOPHARMACEUTICSDocument169 pagesBIOPHARMACEUTICSFitria Shizuoka Jiyeon100% (5)
- Biopharmaceutics and Pharmacokientics A Pracitcal Manual Compiled by Deepa MKDocument76 pagesBiopharmaceutics and Pharmacokientics A Pracitcal Manual Compiled by Deepa MKYuppie Raj100% (2)
- Introduction To Pharmacology: Renato I. Dalmacio, RPH Pharmacology 325 College of PharmacyDocument40 pagesIntroduction To Pharmacology: Renato I. Dalmacio, RPH Pharmacology 325 College of PharmacyJon Paulo Bautista100% (3)
- Biopharmaceutics and Pharmacokinetics-A Treatise BrahmankarDocument206 pagesBiopharmaceutics and Pharmacokinetics-A Treatise BrahmankarLakshmi Kumari100% (4)
- Pharmacokinetics - Exam 3 SPR 2012 Answer KeyDocument6 pagesPharmacokinetics - Exam 3 SPR 2012 Answer KeyCharlie BravoNo ratings yet
- Ayurveda Treatment SkinDocument4 pagesAyurveda Treatment SkinAkash SharmaNo ratings yet
- 1 Introduction - BiopharmaceuticsDocument32 pages1 Introduction - BiopharmaceuticsLouie Fernand LegaspiNo ratings yet
- Prepared by Muhammad Salman: This Brand Plan Has Been Designed For The ProductDocument90 pagesPrepared by Muhammad Salman: This Brand Plan Has Been Designed For The ProductMuhammad SalmanNo ratings yet
- Pharmacokinetics CalculationDocument69 pagesPharmacokinetics CalculationHinna Sood100% (3)
- Toxicokinetics DynamicsDocument76 pagesToxicokinetics DynamicsSunil100% (1)
- Toxicokinetic PDFDocument29 pagesToxicokinetic PDFKirush MitaNo ratings yet
- Multiple Dosage RegimenDocument27 pagesMultiple Dosage Regimenpradeep kumar88% (16)
- Pharmakokinetics Problems SolvedDocument17 pagesPharmakokinetics Problems Solvednikhildhargawe50% (4)
- Therapeutic Drug Monitoring: RVS Chaitanya KoppalaDocument33 pagesTherapeutic Drug Monitoring: RVS Chaitanya KoppalaDr. Raghavendra Kumar GundaNo ratings yet
- Multiple Dose RegimenDocument25 pagesMultiple Dose Regimenalexpharm100% (6)
- Biopharmaceutics and PharmacokineticsDocument76 pagesBiopharmaceutics and PharmacokineticsEswar Gupta Maddi100% (4)
- Physical Pharmacy I-II Sem JNTUK Lab ManualDocument75 pagesPhysical Pharmacy I-II Sem JNTUK Lab ManualBabu Vij100% (4)
- Bioavailability & Bioequivalence StudiesDocument45 pagesBioavailability & Bioequivalence StudiesHussein Talal Kenaan100% (2)
- Pharmacokinetics AbsorptionDocument27 pagesPharmacokinetics AbsorptionchondroboraNo ratings yet
- Introduction To Biopharmaceutics and PharmacokineticsDocument29 pagesIntroduction To Biopharmaceutics and PharmacokineticsNasima Begum100% (1)
- Chapter 1 Introduction To Biopharmaceutics and Pharmacokinetics 1Document92 pagesChapter 1 Introduction To Biopharmaceutics and Pharmacokinetics 1Marc Alamo100% (2)
- @MedicalBooksStore 2015 Office Based PDFDocument237 pages@MedicalBooksStore 2015 Office Based PDFGalih WicaksonoNo ratings yet
- Introduction To BiopharmaceuticsDocument27 pagesIntroduction To BiopharmaceuticsAmina Akther Mim 1821179649No ratings yet
- 2.one Compartment Open ModelDocument90 pages2.one Compartment Open ModelBLESSY SARA KURIANNo ratings yet
- Introduction To BiopharmaceuticsDocument34 pagesIntroduction To BiopharmaceuticsMd. Yasir Galib 1721609649100% (1)
- Pharmacokinetic Exercises ListDocument5 pagesPharmacokinetic Exercises ListVijendra R100% (1)
- Therapeutic Drug Monitoring: Saeed Alqahtani, Pharmd, PHDDocument78 pagesTherapeutic Drug Monitoring: Saeed Alqahtani, Pharmd, PHDAnonymous hF5zAdvwCC50% (2)
- VCI MSVE 2008 RegulationsDocument136 pagesVCI MSVE 2008 RegulationsSunil50% (2)
- VPT MCQ ForDocument10 pagesVPT MCQ ForSunilNo ratings yet
- Chapter 3. One-Compartment Open Model Intravenous Bolus AdministrationDocument23 pagesChapter 3. One-Compartment Open Model Intravenous Bolus AdministrationbencleeseNo ratings yet
- Homeopathy and PaediatricsDocument15 pagesHomeopathy and PaediatricsCristinaNo ratings yet
- Drugs in ReptilesDocument71 pagesDrugs in ReptilesSunil100% (1)
- Practical (3+4+5)Document19 pagesPractical (3+4+5)Asma Alenezy50% (2)
- Rab297cen - Yumizen H500 OT Daily Guide PDFDocument42 pagesRab297cen - Yumizen H500 OT Daily Guide PDFZhafira Afsheen Niesa100% (2)
- Clinical PharmacokineticsDocument31 pagesClinical PharmacokineticsArdiyanti Puspitasari100% (1)
- 02 - One Compartment IV BolusDocument79 pages02 - One Compartment IV BolusAna Francisca100% (1)
- CalculationDocument24 pagesCalculationhablet1100% (1)
- Chapter 11 Multiple Dosage RegimenDocument35 pagesChapter 11 Multiple Dosage RegimenYuli Irvaransiah DIatun NIkmah100% (2)
- Compartment ModelingDocument94 pagesCompartment ModelingPinkishBlue100% (1)
- Pharmacy and Therapeutic CommitteeDocument26 pagesPharmacy and Therapeutic CommitteeRana Ehtisham100% (2)
- Non Compartmental PharmacokineticsDocument72 pagesNon Compartmental PharmacokineticsJaspreet Guraya90% (10)
- BioassayDocument38 pagesBioassayMuhammad Masoom AkhtarNo ratings yet
- 01 BiopharmaceuticsDocument69 pages01 BiopharmaceuticsRiczen Mae F. Vila100% (1)
- Application of PK in Clinical SitutionDocument42 pagesApplication of PK in Clinical Situtionsafia mehmood100% (1)
- Pharmacokinetic Model Questions by Shimaji GDDocument22 pagesPharmacokinetic Model Questions by Shimaji GDShemaj Gurchuma100% (1)
- Carbon Monoxide PoisoningDocument22 pagesCarbon Monoxide PoisoningSunilNo ratings yet
- Classification and Dosage of Antimicrobial Agents in Veterinary MedicineDocument24 pagesClassification and Dosage of Antimicrobial Agents in Veterinary MedicineSunil0% (1)
- Biopharmaceutics and Pharmacokinetics 4th Pharm DDocument6 pagesBiopharmaceutics and Pharmacokinetics 4th Pharm DAnanda Vijayasarathy83% (6)
- Dose Adjustment in Renal and Hepatic FailureDocument27 pagesDose Adjustment in Renal and Hepatic Failurevanita100% (1)
- PMDC 3rd - Schedule NotifiedDocument132 pagesPMDC 3rd - Schedule NotifiedMariaLakhaniNo ratings yet
- BioavailabilityDocument16 pagesBioavailabilityTyshanna JazzyNicole BariaNo ratings yet
- Patterson - Allergic Diseases 6th EdDocument370 pagesPatterson - Allergic Diseases 6th Edvasile100% (2)
- Pharmacokinetic ParametersDocument14 pagesPharmacokinetic ParametersPutri Rahma Fanni0% (1)
- Mycotoxins in Animal HealthDocument103 pagesMycotoxins in Animal HealthSunil100% (1)
- Mycotoxins in Animal HealthDocument103 pagesMycotoxins in Animal HealthSunil100% (1)
- Pharmacokinetics Review CEE With Practice ProblemsDocument148 pagesPharmacokinetics Review CEE With Practice ProblemsBenhur Leithold LapitanNo ratings yet
- BioequivalenceDocument30 pagesBioequivalenceTejas PatelNo ratings yet
- Pharamcokinetics: Course In-Charge: Nimra Waheed Course Name: Biopharmaceutics and Pharmacokinetics Course Code: 613-TDocument21 pagesPharamcokinetics: Course In-Charge: Nimra Waheed Course Name: Biopharmaceutics and Pharmacokinetics Course Code: 613-TNeha GulfamNo ratings yet
- Compartment ModellingDocument46 pagesCompartment ModellingBio DataNo ratings yet
- Nonlinear Pharmacokinetics 1Document34 pagesNonlinear Pharmacokinetics 1donndisaster100% (1)
- 5 Biopharmaceutic Considerations of A Drug DesignDocument89 pages5 Biopharmaceutic Considerations of A Drug DesignImDanaBananaaaNo ratings yet
- BASIC PHARMACOKINETICS - CHAPTER 13: Non-Linear KineticsDocument22 pagesBASIC PHARMACOKINETICS - CHAPTER 13: Non-Linear KineticsDrHeba100% (7)
- 2 Conversion of Iv To PoDocument27 pages2 Conversion of Iv To PoSreya Sanil100% (1)
- Bioavailability and BioequivalenceDocument81 pagesBioavailability and BioequivalenceNiharika ModiNo ratings yet
- MDR Repetitive Intravenous InjectionsDocument17 pagesMDR Repetitive Intravenous InjectionsMuhammad MursaleenNo ratings yet
- Pharmacological and Toxicological Screening Methods I (MPL 103T)Document50 pagesPharmacological and Toxicological Screening Methods I (MPL 103T)Sandeep MewadaNo ratings yet
- Key Pharmacokinetic CalculationsDocument10 pagesKey Pharmacokinetic CalculationsMuqaddam Ahmed SalimNo ratings yet
- Fluorosis Phosphorous Nitrate Toxicity in AnimalsDocument55 pagesFluorosis Phosphorous Nitrate Toxicity in AnimalsSunilNo ratings yet
- Endocrine Vety PharmaDocument16 pagesEndocrine Vety PharmaSunilNo ratings yet
- Household Hazards To PetsDocument16 pagesHousehold Hazards To PetsSunilNo ratings yet
- Classification of AmphibiansDocument22 pagesClassification of AmphibiansSunilNo ratings yet
- Drugs in Behavioural Disorders of PetsDocument5 pagesDrugs in Behavioural Disorders of PetsSunilNo ratings yet
- Toxicological Investigation and Its Significance in Animal Health DiagnosisDocument8 pagesToxicological Investigation and Its Significance in Animal Health DiagnosisSunilNo ratings yet
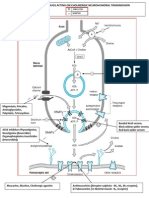
- Site of Action of Drugs Acting On Cholinergic Neurohumoral Transmission PDFDocument1 pageSite of Action of Drugs Acting On Cholinergic Neurohumoral Transmission PDFSunilNo ratings yet
- Antiseptics and Disinfectants For Veterinary ClinicsDocument3 pagesAntiseptics and Disinfectants For Veterinary ClinicsSunil100% (5)
- GENERAL LINE of Treatment UREA AMMONIA SALT - poISONINGDocument49 pagesGENERAL LINE of Treatment UREA AMMONIA SALT - poISONINGSunil0% (1)
- Drugs Acting On Haematopoietic System of AnimalsDocument28 pagesDrugs Acting On Haematopoietic System of AnimalsSunil100% (1)
- Nsaids and Other Antinflammatory Agents in Veterinary PracticeDocument44 pagesNsaids and Other Antinflammatory Agents in Veterinary PracticeSunil100% (2)
- Behavioural Modifying Drugs in PetsDocument33 pagesBehavioural Modifying Drugs in PetsSunilNo ratings yet
- Drugs Acting On Respiratory System of AnimalsDocument8 pagesDrugs Acting On Respiratory System of AnimalsSunil100% (4)
- ZOOTOXINSDocument72 pagesZOOTOXINSSunil83% (6)
- LATHYRISM AND PHOTOSENSiTIZATIONDocument33 pagesLATHYRISM AND PHOTOSENSiTIZATIONSunilNo ratings yet
- Drugs Acting On Genitourinary System of AnimalsDocument44 pagesDrugs Acting On Genitourinary System of AnimalsSunilNo ratings yet
- Environmental PollutantsDocument32 pagesEnvironmental PollutantsSunilNo ratings yet
- Fluorosis Phosphorous ToxicityDocument55 pagesFluorosis Phosphorous ToxicitySunilNo ratings yet
- Site of Action of Drugs Acting On Adrenergic Neurohumoral Transmission PDFDocument1 pageSite of Action of Drugs Acting On Adrenergic Neurohumoral Transmission PDFSunilNo ratings yet
- Blood Vessels and Circulation 1Document36 pagesBlood Vessels and Circulation 1Kuya RnJNo ratings yet
- Pharma Biotech M A Transactions 2005-2012Document13 pagesPharma Biotech M A Transactions 2005-2012Aniket ApteNo ratings yet
- Test 6 Question BankDocument43 pagesTest 6 Question Bankcannon butlerNo ratings yet
- Observator Ios 2016Document355 pagesObservator Ios 2016ajgamesNo ratings yet
- Family Case Study On The Billones Family 1Document63 pagesFamily Case Study On The Billones Family 1Ivy Mae DecenaNo ratings yet
- Msds Nano PolishDocument5 pagesMsds Nano PolishGan LordNo ratings yet
- 220200320sathiya PrakashDocument95 pages220200320sathiya Prakashpriya selvaraj100% (1)
- Branches of ZoologyDocument3 pagesBranches of ZoologyVivek Morya100% (1)
- Final Announcement - 6721760944 PDFDocument23 pagesFinal Announcement - 6721760944 PDFthifla farhaniNo ratings yet
- Final Abstract and Paper Edit 26 April 2020 อ เปรม แก้แล้ว PDFDocument22 pagesFinal Abstract and Paper Edit 26 April 2020 อ เปรม แก้แล้ว PDFchanakarn Vipusmith100% (2)
- PD 03-2020 PDFDocument2 pagesPD 03-2020 PDFtimvrghs123No ratings yet
- Pharmacology Question Bank BPTDocument5 pagesPharmacology Question Bank BPTJoHN SamueLNo ratings yet
- Alatrol: Cetirizine Hydrochloride BPDocument1 pageAlatrol: Cetirizine Hydrochloride BPAfsana AfrinNo ratings yet
- WEB VTH 01292009Document28 pagesWEB VTH 01292009Dave L100% (1)
- Music Therapy Improves Sleep Quality in Acute and ChronicDocument12 pagesMusic Therapy Improves Sleep Quality in Acute and ChronicLaras Ciingu SyahrezaNo ratings yet
- Prob PDFDocument12 pagesProb PDFwallace120No ratings yet
- 1998-08 HP JournalDocument90 pages1998-08 HP JournalElizabeth WilliamsNo ratings yet
- Medication BookDocument448 pagesMedication BookAlbert ChuwaNo ratings yet
- Assessing Breasts and AxillaeDocument23 pagesAssessing Breasts and AxillaeKamile Abrahan GarciaNo ratings yet
- Drug To Drug InteractionDocument47 pagesDrug To Drug InteractionMannan SokaNo ratings yet
- Moretsu Shain (Fanatical Workers) and Yoi Kigyo Senshi (Good Corporate Soldiers)Document7 pagesMoretsu Shain (Fanatical Workers) and Yoi Kigyo Senshi (Good Corporate Soldiers)Erwin Alvih Taufik HidayatNo ratings yet
- Peran Perawat Dalam Penanggulangan Bencana: Vol 6, No 1 Mei, Pp. 63-70 P-ISSN 2549-4880, E-ISSN 2614-1310 WebsiteDocument8 pagesPeran Perawat Dalam Penanggulangan Bencana: Vol 6, No 1 Mei, Pp. 63-70 P-ISSN 2549-4880, E-ISSN 2614-1310 WebsiteEbato SanaeNo ratings yet